 Patrícia Martins Simões1,2,3*Hajar Lemriss4Yann Dumont5Sanâa Lemriss6Jean-Philippe Rasigade2,3Sophie Assant-Trouillet1,2Azeddine Ibrahimi4Saâd El Kabbaj6Marine Butin1,7Frédéric Laurent1,2,3
Patrícia Martins Simões1,2,3*Hajar Lemriss4Yann Dumont5Sanâa Lemriss6Jean-Philippe Rasigade2,3Sophie Assant-Trouillet1,2Azeddine Ibrahimi4Saâd El Kabbaj6Marine Butin1,7Frédéric Laurent1,2,3- 1Department of Clinical Microbiology, Northern Hospital Group, Hospices Civils de Lyon, Lyon, France
- 2International Centre for Research in Infectious Diseases, Institut National de la Santé et de la Recherche Médicale U1111, University of Lyon, Lyon, France
- 3National Reference Center for Staphylococci, Hospices Civils de Lyon, Lyon, France
- 4Biotechnology Laboratory (Medbiotech), Medical and Pharmacy School, University Mohammed V de Rabat, Rabat, Morocco
- 5Department of Clinical Microbiology, Eastern Hospital Group, Hospices Civils de Lyon, Lyon, France
- 6Department of Biosecurity PCL3, Laboratory of Research and Medical Analysis of the Fraternal of Gendarmerie Royale, Rabat, Morocco
- 7Neonatal Intensive Care Unit, Eastern Hospital Group, Hospices Civils de Lyon, Lyon, France
The multi-resistant Staphylococcus capitis clone NRCS-A has recently been described as a major pathogen causing nosocomial, late-onset sepsis (LOS) in preterm neonates worldwide. NRCS-A representatives exhibit an atypical antibiotic resistance profile. Here, the complete closed genome (chromosomal and plasmid sequences) of NRCS-A prototype strain CR01 and the draft genomes of three other clinical NRCS-A strains from Australia, Belgium and the United Kingdom are annotated and compared to available non-NRCS-A S. capitis genomes. Our goal was to delineate the uniqueness of the NRCS-A clone with respect to antibiotic resistance, virulence factors and mobile genetic elements. We identified 6 antimicrobial resistance genes, all carried by mobile genetic elements. Previously described virulence genes present in the NRCS-A genomes are shared with the six non-NRCS-A S. capitis genomes. Overall, 63 genes are specific to the NRCS-A lineage, including 28 genes located in the methicillin-resistance cassette SCCmec. Among the 35 remaining genes, 25 are of unknown function, and 9 correspond to an additional type I restriction modification system (n = 3), a cytosine methylation operon (n = 2), and a cluster of genes related to the biosynthesis of teichoic acids (n = 4). Interestingly, a tenth gene corresponds to a resistance determinant for nisin (nsr gene), a bacteriocin secreted by potential NRCS-A strain niche competitors in the gut microbiota. The genomic characteristics presented here emphasize the contribution of mobile genetic elements to the emergence of multidrug resistance in the S. capitis NRCS-A clone. No NRCS-A-specific known virulence determinant was detected, which does not support a role for virulence as a driving force of NRCS-A emergence in NICUs worldwide. However, the presence of a nisin resistance determinant on the NRCS-A chromosome, but not in other S. capitis strains and most coagulase-negative representatives, might confer a competitive advantage to NRCS-A strains during the early steps of gut colonization in neonates. This suggests that the striking adaptation of NRCS-A to the NICU environment might be related to its specific antimicrobial resistance and also to a possible enhanced ability to challenge competing bacteria in its ecological niche.
Introduction
Coagulase-negative staphylococci (CoNS) are common commensals of the human skin and mucosa and are also opportunistic pathogens responsible for infections associated with indwelling medical devices and bloodstream infections (Otto, 2009). Among CoNSs, Staphylococcus epidermidis is the most prevalent species and is classically involved in nosocomial bacteremia in patients with co-morbidities including very low-birth weight preterm infants (Otto, 2009). However, the Staphylococcus capitis has also been incriminated in sepsis throughout the world in neonatal intensive care units (NICUs) (Van Der Zwet et al., 2002; Venkatesh et al., 2006; D'mello et al., 2008; Rasigade et al., 2012; Boghossian et al., 2013; Cui et al., 2013). We recently reported that a group of closely related multidrug-resistant (MDR) S. capitis strains belonging to the same clone, named NRCS-A, were present in most French NICUs but absent in the other pediatric or adult ICUs (Rasigade et al., 2012). In subsequent work, we demonstrated the dissemination of NRCS-A strains in distant NICUs throughout the world (Butin et al., 2016). This dissemination is of major concern because of the extensive drug resistance of NRCS-A as well as its ability to become highly endemic in some NICUs, representing up to 40% of all bacteremia cases (Stoll et al., 2002, 2005; Rasigade et al., 2012). Due to the presence of the type V-related staphylococcal chromosome cassette mec (SCCmec), all S. capitis NRCS-A strains are resistant to beta-lactams and exhibit reduced susceptibility to other antimicrobial agents in common use in NICUs, including resistance to aminoglycosides and resistance or heteroresistance to vancomycin (Venkatesh et al., 2006; Rasigade et al., 2012). Overall, the worldwide diffusion of a multidrug-resistant S. capitis clone that is highly adapted to NICU antibiotic selective pressure raises questions about the potential genetic support of its epidemiological success.
Few genome sequences of S. capitis are publicly available to date. In addition, only a single study using the genome of a S. capitis isolate collected from a bloodstream infection in an adult has reported the genomic comparison of virulence factors between S. capitis and S. epidermidis (Cameron et al., 2015). Considering the specific importance NRCS-A in the NICU setting and its international dissemination, we present here the first closed genome sequence of the French NRCS-A prototype strain CR01, a comparison of the whole-genome sequences (WGS) of three other NRCS-A strains collected from distant countries (Australia, Belgium, the United Kingdom), and a description of the genomic features of this NRCS-A clone.
Methods
Genome Sequencing and Assembly
The four strains of S. capitis, CR01, CR03, CR04, and CR05, used in this study were part of a previously published collection, with all strains being of the international S. capitis NRCS-A clone (Butin et al., 2016). The strains were isolated from blood cultures of preterm infants diagnosed with neonatal sepsis in Neonatal Intensive Care Units (NICUs) from four different countries: France (isolate CR01), Belgium (isolate CR03), Australia (isolate CR04) and the United Kingdom (isolate CR05) (for further details see Supplementary Data).
Initially, whole-genome pyrosequencing (454 Life Sciences/Roche) was performed for all 4 isolates, as previously reported (Lemriss et al., 2014, 2015). Single-molecule real-time (SMRT) sequencing was then performed for strain CR01 to obtain a closed reference genome for this clone (Supplementary Data).
Sequencing for de novo assembly was performed using PacBio RS II (Menlo Park, CA, USA). High molecular weight DNA was sheared in a Covaris g-TUBE (Covaris, Woburn, MA, USA) to obtain 20 kb fragments. After shearing, the DNA size distribution was checked using a Fragment Analyzer (Advanced Analytical Technologies, Ames, IA, USA), and 5 μg of the sheared DNA was used to prepare a SMRTbell library with PacBio SMRTbell Template Prep Kit 1 (Pacific Biosciences, Menlo Park, CA, USA) according to the manufacturer's recommendations. The resulting library was size selected using a BluePippin system (Sage Science, Inc. Beverly, MA, USA) for molecules larger than 11 kb. The recovered library was sequenced using 1 SMRT cell with P6-C4 chemistry and MagBeads with a PacBio RSII system (Pacific Biosciences, Menlo Park, CA, USA) at 240 min movie length.
Based on the SMRT cell sequencing, we generated 50,876 post-filter polymerase reads with an average read length of 15.6 kb and an N50 read length of 21.1 kb. The average coverage was 265X; for de novo assembly, the PacBio module “RS_HGAP_Assembly.2” in SMRTpipe version v2.3.0 was used for continuous long reads (CLR) after polishing and error correction with Quiver, as described previously (Chin et al., 2013). DNA methylation was determined using the RS_Modification_and_Motif_Analysis protocol within SMRT Portal v2.30, with a standardized in silico false positive error of ~1%. Only motifs with a mean modification quality value (QV) >50 and a mean coverage of >100X were validated as being modified (https://github.com/PacificBiosciences/Bioinformatics-Training/wiki/Methylome-Analysis-Technical-Note). Two polished contigs, one corresponding to the chromosome and the other to the plasmid, were obtained. Circularization of both contigs was achieved by manual comparison and removal of regions of overlaps.
Computational Analysis
Automatic syntactic and functional annotation of the closed, reference genome of strain CR01 [EMBL accession number: LN866849 (chromosome) and LN866850 (plasmid)] was performed using the MicroScope platform pipeline (Vallenet et al., 2006, 2013). The annotations of the draft genomes of strains CR03 (EMBL accession number: CVUF01000001-CVUF01000031), CR04 (EMBL accession number: CTEM01000001-CTEM01000038) and CR05 (EMBL accession number: CTEO01000001-CTEO01000039) were also performed using the MicroScope platform pipeline, as published previously (Lemriss et al., 2014). Visual inspection and manual curation were carried out using the MaGe platform and BLAST searches against NCBI databases.
Virulence factors were identified using the Virulence Factors database (VFDB; http://www.mgc.ac.cn/VFs/, Chen et al., 2012) and tblastn (using the ncbi-blast-2.2.27+ suite, Camacho et al., 2009) searches against in-house databases of known virulence genes of staphylococci.
Prophage regions were identified using the PHAST server (http://phast.wishartlab.com, Zhou et al., 2011). The predicted results were confirmed using the MaGe platform.
For comparison, we obtained publicly available assemblies for six additional whole-genome sequences of S. capitis: strains QN1 (NCBI Ref. Seq.: NZ_AJTH00000000.1), VCU116 (NCBI Ref. Seq.: NZ_AFTX00000000.1), SK14 (NCBI Ref. Seq.: NZ_ACFR00000000.1), LNZR-1 (NCBI Ref. Seq.: NZ_JGYJ00000000.1), C87 (NCBI Ref. Seq.: NZ_ACRH00000000.1), and AYP1020 (NCBI Ref. Seq.: NZ_CP007601.1). All six genomes were aligned against the closed genome of isolate CR01 using Mauve Progressive v.2.3.1 (Darling et al., 2010) with default settings.
Resistance genes were identified by a combination of ResFinder v.2.1 (Kleinheinz et al., 2014) and tblastn searches using the complete, curated Antibiotic Resistance Proteins database (multifasta file) available at the Comprehensive Antibiotic Resistance Database (CARD) (McArthur et al., 2013). For ResFinder, cutoffs of 80% minimum length and 90% identity were used to search for resistance genes in CR03, CR04, and CR05 assemblies previously performed against the reference CR01 using Mauve Progressive to determine their location and genomic context. For detection of resistance genes using the CARD complete, curated Antibiotic Resistance Proteins database, tblastn searches against a database composed of the four genomes of strains CR01, CR03, CR04, and CR05 were performed.
Genes specific to the NRCS-A clone were identified using the MaGe platform “Gene Phyloprofiler” tool (Vallenet et al., 2013) and the four annotated genomes of NRCS-A and public S. capitis genomes. Briefly, genomes were compared in terms of gene content using pre-computed homologies and synteny groups. Pairwise comparisons between predicted protein sequences of the studied genome and the proteins of another genome allowed computation of ranked hits and determination of bidirectional best hits (BBH) (for each protein, the three best hits were retained). Putative orthologous relationships between two genomes were defined as gene couples satisfying the BBH criterion and an alignment threshold for homology set as a minimum of 50% sequence identity along 80% of the length of the smallest protein. Mauve alignments were also used to complement the genomic context, followed by manual curation of the putative genes exclusively found in only the four NRCS-A genomes.
Insertion sequences (IS) in the genomes were identified using the ISfinder analysis tool (Siguier et al., 2006). Both blastn and blastp queries were performed with default settings. Matches with an e-value smaller than 0.5 were manually curated. To confirm whether a given IS position was conserved in the other genomes analyzed, the closed genome of isolate CR01 was aligned with the three other NRCS-A genomes and with the six public genomes using Mauve Progressive v.2.3.1 (Darling et al., 2010) with default settings. ORFs within immediate proximity of conserved IS loci in the NRCS-A genomes were compared with the S. capitis reference genome to check for potential alterations in coding sequences, using both Mauve Progressive and Mage platform. genomic islands (GIs) were predicted using IslandViewer 3 (Dhillon et al., 2015) and confirmed by alignment with the three NCRS-A genomes and the six public S. capitis genomes using Mauve Progressive. IS insertions within GIs were predicted as mentioned above. NRCS-A-specific GIs were searched in the NCBI database using megablast (Camacho et al., 2009) query, limiting to the Staphylococcus taxon (taxid: 1279). Published GIs from reference genomes were searched against Staphylococcus aureus Mu50 (NCBI Reference Sequence: NC_002758.2), COL (NCBI Reference Sequence: NC_002951.2), MW2 (NCBI Reference Sequence: NC_003923.1), N315 (NCBI Reference Sequence: NC_002745.2), FPR3757 (NCBI Reference Sequence: NC_007793.1) and S0385 (NCBI Reference Sequence: NC_017333.1), and S. epidermidis RP62A (NCBI Reference Sequence: NC_002976.3) using the Mage platform [15]. Syntonomes of more than 3 ORFs and with identity greater than 33% were considered to be GIs.
Investigation of nsr Gene Presence in Clinical Bacterial Strains
A panel of 15 strains (Table 1) comprising clinical strains of S. capitis (NRCS-A and non-NRCS-A) were used for nisin susceptibility testing. All staphylococcal species were originally isolated from blood, and species assignment was determined using Matrix-Assisted Laser Desorption/Ionization Time of Flight Mass Spectrometry (MALDI-TOF Vitek MS, Biomerieux, France). After isolation, the strains were cultured on Horse Blood Agar (Biomerieux) and then stored at −80°. Testing for susceptibility to nisin was performed using the disk diffusion test adapted from EUCAST1. Briefly, nisin solutions were prepared using 2.5% nisin powder (Sigma Aldrich, USA) according to a previously described protocol (Piper et al., 2009). Sterile paper disks (Thermofisher Diagnostics, France) were soaked with 30 μg of nisin solution. After an overnight incubation at 36°C on Horse blood Agar (Biomerieux, France), a 0.5 McF was seeded on Mueller Hinton agar (MHE) plates (Biomerieux, France). A disk containing 30 μg of nisin was placed in the middle of the plate. The diameter of the inhibition zones was measured after overnight incubation of the inoculated MHE plates at 36°C. The average diameter of inhibition and SEM were determined from four independent experiments.
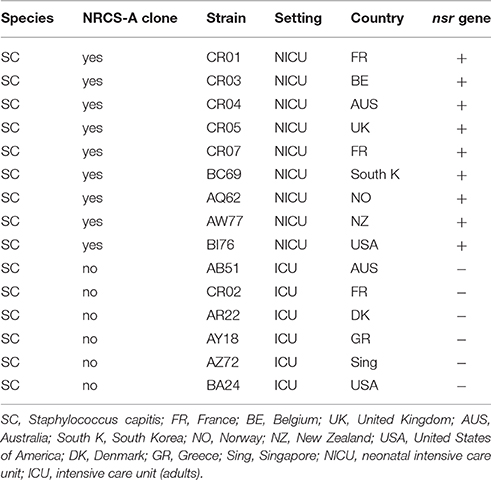
Table 1. Bacterial strains used in the nisin susceptibility test.
PCRs for detecting the nsr gene were performed using consensus primers: pF–word-5′ GGAGATATGGGACCTATGATTGCA 3′ and pR–word-5′ GCTGTAAkTTCwCCkGAACTkGC 3′. These primers were designed based on alignment of the nsr gene sequence found in strain CR01 (S. capitis clone NRCS-A) and sequences retrieved from NCBI after a blastn (ref) search for homologous sequences in staphylococcal WGS genomes [S. hyicus ATCC 11249 (acc. num. CP008747), Staphylococcus sp. TE8 (acc. num. NZ_JMGB00000000.1), S. epidermidis MC28 (acc. num. NZ_ATCZ00000000.2), S. epidermidis VCU128 (acc. num. NZ_AHLI00000000.1)]. PCR amplification was conducted in a final volume of 25 μL, with 2.0 μL bacterial DNA (QuickExtract kit, Qiagen), 2.5 μL 10x Reaction Buffer, 0.75 μL 50 mM MgCl2, 1 μL 10 mM forward primer, 1 μL 100 mM reverse primer, 4 μL 5 mM dNTPs, 1 μL each primer and 0.125 μL Taq polymerase (Eurobio, France). The cycling parameters were as follows: (i) initial denaturation for 3 min at 95°C; (ii) 34 cycles, with 1 cycle consisting of 30 s at 95°C, 60 s at 60°C, and 90 s at 72°C; and (iii) a final extension for 10 min at 72°C.
Results
WGSs of four strains belonging to S. capitis clone NRCS-A and originating from NICUs in four distant countries were obtained to investigate lineage-specific virulomes, resistomes, and mobilomes as well as the presence of unshared genes using 454 pyrosequencing technology, as reported previously (Lemriss et al., 2014, 2015). Depending on the isolate, assembly of the draft genomes produced 26 to 39 contigs. Re-sequencing of strain CR01 using SMRT technology (PacBio) allowed for the generation of closed complete sequences for the chromosome and the single plasmid that was subsequently used to produce scaffolds for the three remaining WGSs.
The length of CR01's chromosome is 2,522,871 bp and exhibits a low G + C content (33.02%), as is expected for staphylococci. We identified and annotated 2419 protein-coding regions, 19 rRNAs, 62 tRNAs, 34 ncRNAs (including RNAIII), and 20 transposases associated with insertion sequence elements and transposons. Of note, gaps (identified in the draft genome assemblies of the four NRCS-A isolates (based on 454 pyro-sequencing)) frequently occur within the vicinity of transposases, suggesting that some of the 454 short read assemblies were unable to bridge the gaps associated with repetitive genomic elements. These data clearly emphasize the suitability of third-generation sequencing technologies, such as PacBio SMRT, for obtaining fully defined de novo assemblies and, thus, for overcoming the issue of both local and global repeats (Koren and Phillippy, 2015). The genomic characteristics of the NRCS-A isolates tested in this study are summarized in Table 2.

Table 2. General genomic features of S. capitis strain CR01 compared with another three WGS NRCS-A genomes.
Virulome
The presence of virulence-associated genes in the closed genome of strain CR01 and subsequently the draft genomes of strains CR03 (Be), CR04 (Aus), and CR05 (UK) were inferred by comparison with known virulence factors previously reported for S. aureus, S. epidermidis, and S. haemolyticus using the VFDB database (Chen et al., 2012). The findings were augmented with BLAST searches of well-characterized staphylococcal virulence factors and key regulators (Table 3). Comparison of the four NRCS-A genomes with the six published genomes of non-NRCS-A S. capitis, including QN1, VCU116, SK14, LNZR-1, C87, and AYP1020 strains, showed that none of the known staphylococcal virulence genes are exclusively carried by the NRCS-A clone.
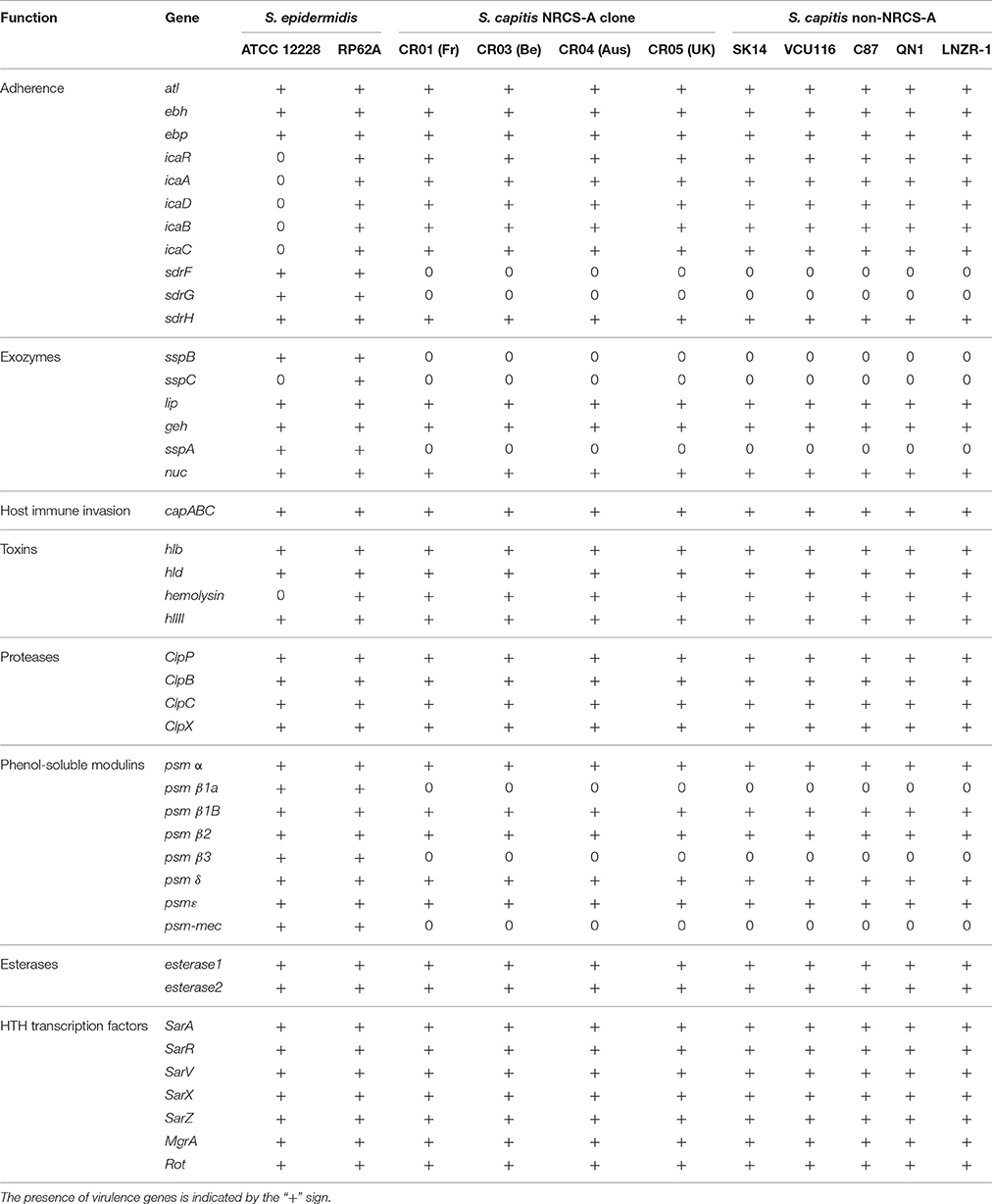
Table 3. Comparison of virulence factors in S. capitis NRCS-A, non-NRCS-A, and S. epidermidis after exclusion of virulence factors present only in S. aureus.
Interestingly, the virulome of the NRCS-A S. capitis genome was quite similar to that of S. epidermidis RP62A, including the icaABDCR and capABC biofilm-related operons, Clp proteases and multiple copies of PSM beta type 1b in tandem (75% aa identity to PSMbeta1b of strain RP62) and one PSM alpha (59% aa identity) (Otto, 2012). All PSMs were found in the same genetic environment as in the S. epidermidis RP62A genome, with conservation of upstream and downstream genes.
As previously observed for the fully closed S. epidermidis RP62a and S. epidermidis ATCC 12228 genomes (Zhang et al., 2003; Gill et al., 2005), no S. aureus toxins, except beta- and delta-hemolysins, were identified in any of the 10 S. capitis genomes (four NRCS-A and six public non-NRCS-A). Similarly, none of the genes associated with secretion systems (esxA, esxB, esaA, esaB, esaC, essA, essB, and essC) or S. aureus serine proteases (splA, B, C, D, E, and F) are present in the S. capitis genomes.
Resistome
Several antibiotic resistance-associated genes were identified and correlated with the specific resistance phenotype previously reported for the NRCS-A clone (Rasigade et al., 2012; Table 4). All are located on mobile genetic elements (MGEs). Resistance to aminoglycosides is related to the bifunctional aminoglycoside-modifying gene aacA-aphD, which is carried on the transposon Tn4001 (GenBank accession no. AB682805.1; Lyon et al., 1984). Resistance to methicillin is due to the presence of a composite SCCmec-SCCcad/ars/cop cassette exclusively present in the NRCS-A clone, as recently published by our team (Martins Simões et al., 2013). Genomic comparison between the four NRCS-A strains showed that this mobile element is nearly identical: all ORFs are conserved (100% aa homology), and the few nucleotide sequence variations occur only in the CRISPR repeat region, as is expected for these regions. As previously reported (Martins Simões et al., 2013), no other antibiotic resistance genes were detected for this composite SCC element. Finally, the plasmidic bla operon, coding for blaZ beta-lactamase and its regulators, is present in all NRCS-A strains, except strain CR05 (UK).
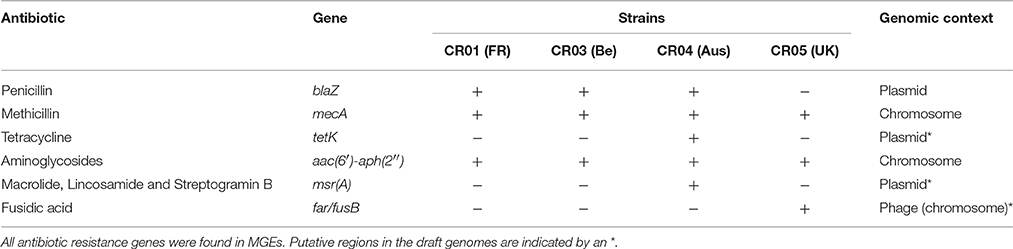
Table 4. Genomic profiles of antibiotic resistance for clone NRCS-A strains from four different countries.
With regard to other antimicrobial families, for which variability in resistance profiles have been observed among NRCS-A isolates, phenotypic antimicrobial susceptibility testing matched the specific resistance gene contents of each NRCS-A strain. Plasmidic msrA and tetK genes, respectively involved in erythromycin resistance (with a negative D-test) and tetracycline resistance, were only identified in strain CR04 (Australia). In strain CR05 (UK), the gene far1, which is responsible for fusidic acid resistance, is carried by a putative phage.
Mobile Elements
As mentioned previously, the shortest circular sequence obtained by SMRT for strain CR01 corresponds to a plasmid of 26.140 bp (GC content of 29.25%). It harbors 35 ORFs, including (i) the bla operon coding for penicillinase resistance to penicillins (see above) and (ii) copper resistance-related genes such as copZ, copA, and csoR (copper transcriptional repressor; Figure 1). Genome comparison of strain CR01's plasmid with the three other NRCS-A draft genomes revealed that the plasmid is not conserved in all NRCS-A strains (Figure 1). In strain CR03 (Belgium), one contig presents 31 of the 35 ORFs identified on the CR01 plasmid. Conversely, in strain CR04 (Australia), only 9 of the 35 ORFs (corresponding to the blaZRI operon and copper resistance operon) were detected in a contig corresponding to a putative plasmid. Finally, no putative plasmid was detected in strain CR05 (UK).
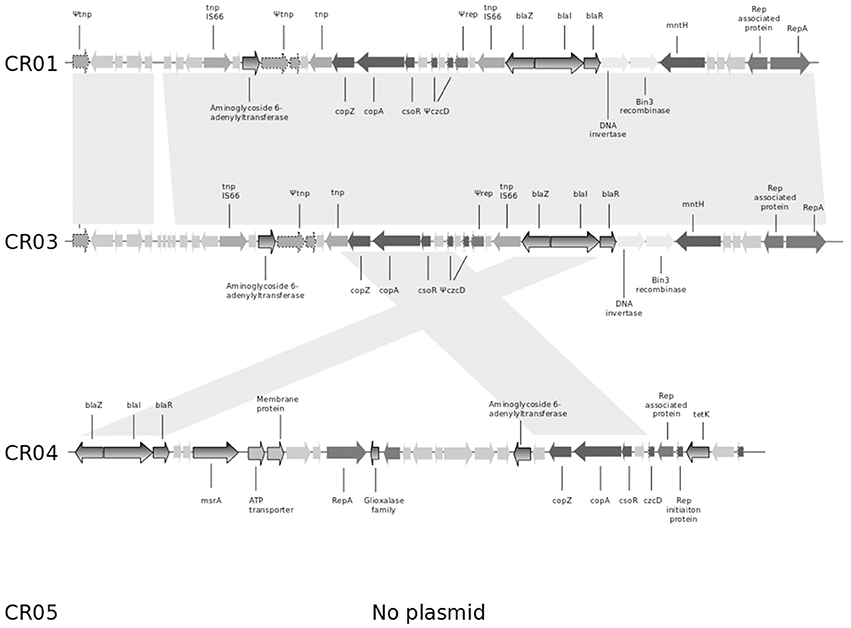
Figure 1. Comparison of the genetic content of the strain CR01 plasmid with the draft genomes of the other three NRCS-A strains. Open reading frames (ORFs) are shown as arrows indicating the direction of transcription. Homologous gene clusters are indicated by a light gray shadow connecting ORFs present in distinct plasmid sequences. Light gray arrows represent unknown proteins, unless specified otherwise. Arrows with dotted lines represent partial or truncated ORFs. Antibiotic resistance genes are colored with a gray gradient; genes associated with resistance to heavy metals are colored in dark gray. Abbreviations: tnp, transposase; copZ, copper insertion chaperone and transporter component; copA, copper transporter ATPase; csoR, copper-sensing transcriptional repressor; czcD, potassium/proton-divalent cation antiporter; rep, replication-associated family protein; repA, replication-associated protein RepA; blaZ, beta-lactamase resistance gene; blaR, regulatory protein BlaR1; blaI, penicillinase repressor; mntH, divalent metal cation transporter MntH, tetK, tetracycline resistance gene.
One intact prophage region was identified in all four NRCS-A genomes. This region shows 100% aa identity to the genomes of strains CR01, CR03, and CR04, but it is degenerate in CR05 (Figure 2). Moreover, this phage exhibits strong homology to a phage present in strain VCU116 [57.1, 90.2, 76.2% aa homology for regions 1, 2 and 3, respectively (Figure 2)] and less than 30% homology with phage Staphy_StB20_(NCBI Ref. Seq. NC_019915), the closest phage found in the NCBI database. Of note, two additional incomplete prophage regions were also detected in strain CR05.
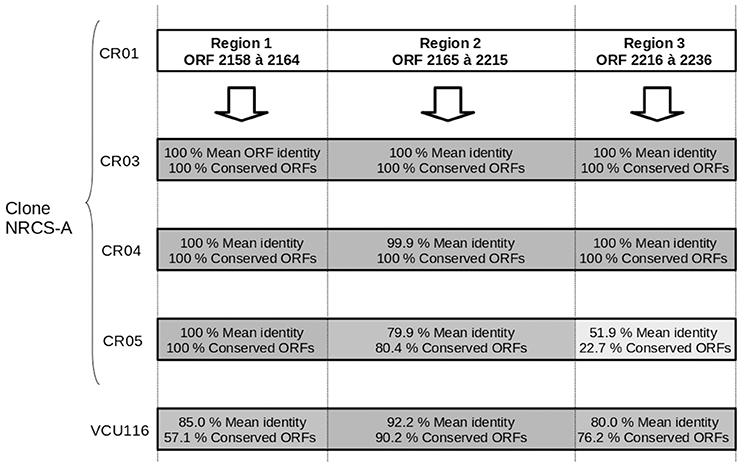
Figure 2. Comparison of the genetic content of the strain CR01 intact prophage present in the other three draft genomes of NRCS-A strains. Prophage prediction was performed using PHAST software; cR01's prophage is divided into three regions according to the degree of conservation compared to the other strains. The mean percent of aa identity in each region is indicated, and a gray gradient is also used to represent the percent similarity.
Nineteen distinct insertion sequence (IS) elements, clustering into three distinct families (type IS256, n = 4; type IS1272, n = 5; IS431mec, n = 10), were identified in the CR01 genome. The presence of type IS256 was confirmed in all four NRCS-A strains, with two IS256 elements being associated with the aminoglycosides resistance gene aacA-aphD (′aac(6′)-aph(2″) on transposon Tn4001. Type IS1272 was also found in strains CR03 and CR04 but not in strain CR05, and their genomic locations are not conserved. Two IS431mec cassettes are present in all NRCS-A genomes, as they are carried within the SCCmec-SCCcad/ars/cop element that is specific to and conserved among the NRCS-A lineage (Martins Simões et al., 2013).
Finally, other GIs (n = 5) were predicted for CR01 (Table 2) and found in the CR03, CR04, and CR05 strains. However, only one was found exclusively in the NRCS-A lineage. This GI (bp position: 150,780–158,140 in the strain CR01 genome) contains six ORFs: one putative phosphoglycolate phosphatase, one conserved protein of unknown function and three pseudogenes of phage proteins (bp position: 151,470–152,238). Unexpectedly, two of these pseudogenes present 40 and 46.5% aa identity to Listeria monocytogenes phage B025 protein gp27 and the third pseudogene presents 60.6% aa identity to L. monocytogenes phage B025 protein gp28. Both gp27 and gp28 proteins have unknown functions.
Methylome and Restriction Modification Systems
SMRT technology (PacBio) allows for genome-wide detection of modified nucleotides based on the rate at which DNA polymerase incorporates bases during sequencing (Flusberg et al., 2010, Nat. Methods; Roberts et al., 2013). Analysis of polymerase kinetic profiles in strain CR01 identified 1333 methylated positions (Table S2) including 94% (n = 1253) corresponding to adenine methylations (m6A) and 0.006% (n = 8) to cytosine methylations (m4C). The adenine modifications observed correlate with the presence of two adenine restriction-modification systems (hsdMSR 1 pos: 476,473–482,360 bp; hsdMSR 2 pos: 686,750–691,866 bp) in the strain CR01 genome, whereas the presence of an mcrBC 5-methylcytosine restriction system (bp position 487,030–492,276) correlates with the cytosine modifications detected. Interestingly, the latter is associated in tandem with the specific additional hdsMSR 1 operon located immediately after the SCCmec-SCCcad/ars/cop element (see above).
NRCS-A Clone Specific Genes
Comparison of the closed genome of strain CR01 with the draft genomes of the three other NRCS-A genomes and with the six public non-NRCS-A genomes revealed a unique set of 63 genes present exclusively in the NRCS-A clone genome (Table S1). Of these, 28 ORFS are carried by the composite SCCmec-SCCcad/ars/cop mobile element, which is related to the acquisition of methicillin resistance (mecA gene). Interestingly, within this cassette, the CRISPR element is specific to the NRCS-A lineage (Martins Simões et al., 2013). Of note, both the clone-specific cassette (28 genes) and its genetic environment (downstream and upstream regions) are fully conserved in the four NRCS-A strains, which confirmed that the four isolates belong to the same clonal population, even though they were isolated from distant countries/continents. Indeed, it is highly unlikely that the same SCCmec element (100% identity) could be acquired in four independent events in four distant countries.
Among the 35 remaining genes found exclusively in the NRCS-A lineage, 25 are of unknown function, and 10 correspond to the following: (i) an additional type I restriction modification system (hsdMSR, n = 3 genes), (ii) a cytosine methylation operon (mcrBC, n = 2), (iii) a cluster of four genes known to be involved in the biosynthesis of teichoic acids [ispD (2-C-methyl-D-erythritol 4-phosphate cytidylyltransferase), tarJ (ribitol-5-phosphate dehydrogenase), tarK (Glycosyl glycerophosphate), and tagF (CDP-glycerol glycerophosphotransferase), n = 4]. Of note, the tenth gene encodes a 316 aa protein presenting 41% aa homology with the determinant for resistance to nisin (nsr gene) that is present in some Lactococcus lactis strains (Froseth and McKay, 1991; Liu et al., 2005) and which shows protease activity. Contrary to the plasmidic localization of the nsr gene in nisin-resistant L. lactis strains, the nsr gene in the NRCS-A clone is located on the chromosome (position: 521,747–522,694 bp) immediately upstream from the potassium (K+) transporter kdpEDABC operon conserved in all S. capitis isolates and downstream from the four teichoic acid biosynthesis genes (ispD, tarJ, tarK, and tagF; see above). No IS or transposon-like region was identified near the nsr gene.
Phenotypic Assay of Clone NRCS-A Nisin Resistance
To assess functional expression of the nisin resistance gene (nsr) found exclusively in the NRCS-A clone, we performed a disk diffusion test with nisin-charged disks using a collection of S. capitis strains, including nine strains belonging to the NRCS-A clone and six nsr-negative, non-NRCS-A strains. The S. capitis strains belonging to the NRCS-A clone presented significantly lower nisin inhibition zones (mean of four independent measurements ± SEM: 8.81 mm ± 1.31) than the strains isolated from adults that do not carry the nsr gene (16.5 ± 1.5; the Student t-test, p < 0.001 vs. 8.8 mm ± 1.3 in NRCS-A). This confirms that the nsr gene found exclusively in S. capitis strains belonging to the neonatal NRCS-A clone is functional and confers resistance to nisin. Nonetheless, it is known (i) that S. aureus (SA) can modulate its resistance to nisin and other small antibacterial peptides via two-component systems (TCSs) GraSR, NsaSR, BraSR in association with ABC transporters (Howden et al., 2010; Blake et al., 2011; Falord et al., 2011; Kolar et al., 2011; Kawada-Matsuo et al., 2013) and (ii) that SA strains resistant or heteroresistant to vancomycin (VISA and hVISA) usually present increased cell wall thickening (Cui et al., 2003). The same mechanisms have been observed in L. lactis resistant to nisin (Kramer et al., 2006; Shin et al., 2016). Thus, to assess whether the resistance to nisin observed in the NRCS-A strains is due exclusively to expression of the nsr gene and not to a cell wall-thickening adaptation, complementary experiments are required using nsr+/nsr− isogenic isolates. Such analyses are beyond the scope of the present paper.
Discussion
The NRCS-A clone is of major interest due to its worldwide high dissemination in NICUs and its prevalence as an agent of neonatal sepsis in preterm newborns (Butin et al., 2016). Comparison between the complete genome of the prototype strain belonging to the S. capitis NRCS-A clone (strain CR01, France) with the draft genomes of clinical S. capitis isolates belonging or not to the NRCS-A clone revealed the presence of multiple MGEs mediating the atypical antibiotic resistance profile of this clone. These data confirm (i) the ability of the NRCS-A clone to adapt to the specific selective pressure of the antibiotics used in NICUs and (ii) emphasize the ability of WGS to provide accurate predictions of the resistance phenotypes of staphylococci and to become a promising alternative for culture methods.
Based on genomic comparisons using previously characterized staphylococcal virulence genes, the NRCS-A virulome is highly similar to that of other non-NRCS-A S. capitis as well as S. epidermidis RP62A strains, which suggests that the success of this clone in neonates is likely not due to increased virulence. Nonetheless, we identified the exclusive presence of a gene encoding a functional nisin resistance (nsr) gene in the NRCS-A lineage that is seldom found in staphylococci.
Nisin, a 34 aa antimicrobial peptide produced by a group of gram-positive Lactococcus and Streptococcus species (Shin et al., 2016), is active against a wide range of gram-positive bacteria, including staphylococci. Its mode of action is dual, involving (i) inhibition of cell wall biosynthesis (transglycosylation step), as it binds to and sequesters lipid II from its functional location, and (ii) pore formation (Wiedemann et al., 2001; Peschel and Sahl, 2006; Egan et al., 2016). Nisin has been described as a key player of the gut barrier, and it is widely used as food preservative due to its potent bactericidal activity. Moreover, it has been shown that (i) expression of the nsr gene, identified in Streptococcus agalactiae, in L. lactis not producing nisin induces resistance to nisin and (ii) the presence of human nisin-producing lactic acid bacteria in the gut reduces intestinal colonization by vancomycin-resistant enterococci (Millette et al., 2008).
Taken together, these data and our results strongly suggest that nisin resistance might be responsible for the potential increase in the ability of the NRCS-A clone to establish itself as part of the initial microflora of neonates, in which the Lactococcus genus is one of the dominant bacterial taxa (Park et al., 2005; Morelli, 2008). Expression of the NSR peptidase might enable NRCS-A isolates to establish themselves in the gut of neonates and also to colonize it and survive longer than nisin-susceptible bacteria. This may explain the over-representation of these isolates in sepsis processes, either through direct translocation in blood (Taft et al., 2015) or via colonization of indwelling devices. Although it remains unknown how S. capitis NRCS-A strains are able to translocate into the bloodstream of infants, it is hypothesized that the entry point might be the digestive tract, which is immature in very preterm infants (Taft et al., 2015). Further studies are needed to fully characterize the digestive microflora of very low-weight preterm-infants and its potential impact on the selection of and the fitness advantage to NRCS-A isolates.
Author Contributions
PM: work, study design, data analysis, and manuscript preparation. HL: data analysis and manuscript preparation. YD: data analysis, work, and manuscript preparation. SL: work and manuscript preparation. JR, SA, AI, and SE: manuscript preparation. MB and FL: study design and manuscript preparation.
Funding
The present work was financed by the French Ministry of Health and the French Institute for Public Health Surveillance (INVS) - Santé publique France in the framework of the National Reference Center of Staphylococci and by the grant ING20111223510 from the Fondation pour la Recherche Medical (FRM).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Michele Bes and Helene Meugnier at the National Reference Center for Staphylococci in Lyon (France), and our colleagues at the National Reference Center of Staphylococci in Belgium (Olivier Denis), in the United Kingdom (Angela Kearns) and in Australia (Margaret Deighton).
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmicb.2016.01991/full#supplementary-material
Footnotes
1. ^ European Committee on antimicrobial Susceptibility Testing, 2015, http://www.eucast.org/fileadmin/src/media/PDFs/EUCAST_files/Disk_test_documents/Manual_v_5.0_EUCAST_Disk_Test.pdf
References
Blake, K. L., Randall, C. P., and O'Neill, A. J. (2011). In vitro studies indicate a high resistance potential for the lantibiotic nisin in Staphylococcus aureus and define a genetic basis for nisin resistance. Antimicrob. Agents Chemother. 55, 2362–2368. doi: 10.1128/AAC.01077-10
Boghossian, N. S., Page, G. P., Bell, E. F., Stoll, B. J., Murray, J. C., Cotten, C. M., et al. (2013). Late-onset sepsis in very low birth weight infants from singleton and multiple-gestation births. J. Pediatr. 162, 1120–1124, 1124.e1. doi: 10.1016/j.jpeds.2012.11.089
Butin, M., Rasigade, J.-P., Simões, P. M., Meugnier, H., Lemriss, H., Goering, R. V., et al. (2016). Wide geographical dissemination of multiresistant Staphylococcus capitis NRCS-A clone in neonatal intensive-care units. Clin. Microbiol. Infect. 22, 46–52. doi: 10.1016/j.cmi.2015.09.008
Camacho, C., Coulouris, G., Avagyan, V., Ma, N., Papadopoulos, J., Bealer, K., et al. (2009). BLAST+: architecture and applications. BMC Bioinformatics 10:421. doi: 10.1186/1471-2105-10-421
Cameron, D. R., Jiang, J.-H., Hassan, K. A., Elbourne, L. D., Tuck, K. L., Paulsen, I. T., et al. (2015). Insights on virulence from the complete genome of Staphylococcus capitis. Front. Microbiol. 6:980. doi: 10.3389/fmicb.2015.00980
Chen, L., Xiong, Z., Sun, L., Yang, J., and Jin, Q. (2012). VFDB 2012 update: toward the genetic diversity and molecular evolution of bacterial virulence factors. Nucleic Acids Res. 40, D641–D645. doi: 10.1093/nar/gkr989
Chin, C.-S., Alexander, D. H., Marks, P., Klammer, A. A., Drake, J., Heiner, C., et al. (2013). Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat. Methods 10, 563–569. doi: 10.1038/nmeth.2474
Cui, B., Smooker, P. M., Rouch, D. A., Daley, A. J., and Deighton, M. A. (2013). Differences between two clinical Staphylococcus capitis subspecies as revealed by biofilm, antibiotic resistance, and pulsed-field gel electrophoresis profiling. J. Clin. Microbiol. 51, 9–14. doi: 10.1128/JCM.05124-11
Cui, L., Ma, X., Sato, K., Okuma, K., Tenover, F. C., Mamizuka, E. M., et al. (2003). Cell wall thickening is a common feature of vancomycin resistance in Staphylococcus aureus. J. Clin. Microbiol. 41, 5–14. doi: 10.1128/JCM.41.1.5-14.2003
D'mello, D., Daley, A. J., Rahman, M. S., Qu, Y., Garland, S., Pearce, C., et al. (2008). Vancomycin heteroresistance in bloodstream isolates of Staphylococcus capitis. J. Clin. Microbiol. 46, 3124–3126. doi: 10.1128/JCM.00592-08
Darling, A. E., Mau, B., and Perna, N. T. (2010). progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS ONE 5:e11147. doi: 10.1371/journal.pone.0011147
Dhillon, B. K., Laird, M. R., Shay, J. A., Winsor, G. L., Lo, R., Nizam, F., et al. (2015). IslandViewer 3: more flexible, interactive genomic island discovery, visualization and analysis. Nucleic Acids Res. 43, W104–W108. doi: 10.1093/nar/gkv401
Egan, K., Field, D., Rea, M. C., Ross, R. P., Hill, C., and Cotter, P. D. (2016). Bacteriocins: novel solutions to age old spore-related problems? Front. Microbiol. 7:e461. doi: 10.3389/fmicb.2016.00461
Falord, M., Mäder, U., Hiron, A., Débarbouillé, M., and Msadek, T. (2011). Investigation of the Staphylococcus aureus GraSR regulon reveals novel links to virulence, stress response and cell wall signal transduction pathways. PLoS ONE 6:e21323. doi: 10.1371/journal.pone.0021323
Flusberg, B. A., Webster, D. R., Lee, J. H., Travers, K. J., Olivares, E. C., Clark, T. A., et al. (2010). Direct detection of DNA methylation during single-molecule, real-time sequencing. Nat. Methods 7, 461–465. doi: 10.1038/nmeth.1459
Froseth, B. R., and McKay, L. L. (1991). Molecular characterization of the nisin resistance region of Lactococcus lactis subsp. lactis biovar diacetylactis DRC3. Appl. Environ. Microbiol. 57, 804–811.
Gill, S. R., Fouts, D. E., Archer, G. L., Mongodin, E. F., Deboy, R. T., Ravel, J., et al. (2005). Insights on evolution of virulence and resistance from the complete genome analysis of an early methicillin-resistant Staphylococcus aureus strain and a biofilm-producing methicillin-resistant Staphylococcus epidermidis strain. J. Bacteriol. 187, 2426–2438. doi: 10.1128/JB.187.7.2426-2438.2005
Howden, B. P., Davies, J. K., Johnson, P. D., Stinear, T. P., and Grayson, M. L. (2010). Reduced Vancomycin Susceptibility in Staphylococcus aureus, including Vancomycin-Intermediate and Heterogeneous Vancomycin-Intermediate Strains: resistance mechanisms, laboratory detection, and clinical implications. Clin. Microbiol. Rev. 23, 99–139. doi: 10.1128/CMR.00042-09
Kawada-Matsuo, M., Yoshida, Y., Zendo, T., Nagao, J., Oogai, Y., Nakamura, Y., et al. (2013). Three distinct two-component systems are involved in resistance to the class I bacteriocins, Nukacin ISK-1 and nisin A, in Staphylococcus aureus. PLoS ONE 8:e69455. doi: 10.1371/journal.pone.0069455
Kleinheinz, K. A., Joensen, K. G., and Larsen, M. V. (2014). Applying the ResFinder and VirulenceFinder web-services for easy identification of acquired antibiotic resistance and E. coli virulence genes in bacteriophage and prophage nucleotide sequences. Bacteriophage 4:e27943. doi: 10.4161/bact.27943
Kolar, S. L., Nagarajan, V., Oszmiana, A., Rivera, F. E., Miller, H. K., Davenport, J. E., et al. (2011). NsaRS is a cell-envelope-stress-sensing two-component system of Staphylococcus aureus. Microbiol. Read. Engl. 157, 2206–2219. doi: 10.1099/mic.0.049692-0
Koren, S., and Phillippy, A. M. (2015). One chromosome, one contig: complete microbial genomes from long-read sequencing and assembly. Curr. Opin. Microbiol. 23, 110–120. doi: 10.1016/j.mib.2014.11.014
Kramer, N. E., van Hijum, S. A., Knol, J., Kok, J., and Kuipers, O. P. (2006). Transcriptome analysis reveals mechanisms by which Lactococcus lactis acquires nisin resistance. Antimicrob. Agents Chemother. 50, 1753–1761. doi: 10.1128/AAC.50.5.1753-1761.2006
Lemriss, H., Lemriss, S., Martins-Simoes, P., Butin, M., Lahlou, L., Rasigade, J.-P., et al. (2015). Genome sequences of four Staphylococcus capitis NRCS-A isolates from geographically distant neonatal intensive care units. Genome Announc. 3, e00501–e00515. doi: 10.1128/genomeA.00501-15
Lemriss, H., Martins Simões, P., Lemriss, S., Butin, M., Ibrahimi, A., El Kabbaj, S., et al. (2014). Non-contiguous finished genome sequence of Staphylococcus capitis CR01 (pulsetype NRCS-A). Stand. Genomic Sci. 9, 1118–1127. doi: 10.4056/sigs.5491045
Liu, C.-Q., Su, P., Khunajakr, N., Deng, Y.-M., Sumual, S., Kim, W. S., et al. (2005). Development of food-grade cloning and expression vectors for Lactococcus lactis. J. Appl. Microbiol. 98, 127–135. doi: 10.1111/j.1365-2672.2004.02441.x
Lyon, B. R., May, J. W., and Skurray, R. A. (1984). Tn4001: a gentamicin and kanamycin resistance transposon in Staphylococcus aureus. Mol. Gen. Genet. 193, 554–556.
Martins Simões, P., Rasigade, J.-P., Lemriss, H., Butin, M., Ginevra, C., Lemriss, S., et al. (2013). Characterization of a novel composite staphylococcal cassette chromosome mec (SCCmec-SCCcad/ars/cop) in the neonatal sepsis-associated Staphylococcus capitis pulsotype NRCS-A. Antimicrob. Agents Chemother. 57, 6354–6357. doi: 10.1128/AAC.01576-13
McArthur, A. G., Waglechner, N., Nizam, F., Yan, A., Azad, M. A., Baylay, A. J., et al. (2013). The comprehensive antibiotic resistance database. Antimicrob. Agents Chemother. 57, 3348–3357. doi: 10.1128/AAC.00419-13
Millette, M., Cornut, G., Dupont, C., Shareck, F., Archambault, D., and Lacroix, M. (2008). Capacity of human nisin- and pediocin-producing lactic Acid bacteria to reduce intestinal colonization by vancomycin-resistant enterococci. Appl. Environ. Microbiol. 74, 1997–2003. doi: 10.1128/AEM.02150-07
Morelli, L. (2008). Postnatal development of intestinal microflora as influenced by infant nutrition. J. Nutr. 138, 1791S–1795S. Available online at: http://jn.nutrition.org/content/138/9/1791S.full#fn-1
Otto, M. (2009). Staphylococcus epidermidis: the “accidental” pathogen. Nat. Rev. Micro. 7, 555–567. doi: 10.38/nrmicro2182
Otto, M. (2012). Molecular basis of Staphylococcus epidermidis infections. Semin. Immunopathol. 34, 201–214. doi: 10.1007/s00281-011-0296-2
Park, H.-K., Shim, S.-S., Kim, S.-Y., Park, J.-H., Park, S.-E., Kim, H.-J., et al. (2005). Molecular analysis of colonized bacteria in a human newborn infant gut. J. Microbiol. 43, 345–353.
Peschel, A., and Sahl, H.-G. (2006). The co-evolution of host cationic antimicrobial peptides and microbial resistance. Nat. Rev. Microbiol. 4, 529–536. doi: 10.1038/nrmicro1441
Piper, C., Draper, L. A., Cotter, P. D., Ross, R. P., and Hill, C. (2009). A comparison of the activities of lacticin 3147 and nisin against drug-resistant Staphylococcus aureus and Enterococcus species. J. Antimicrob. Chemother. 64, 546–551. doi: 10.1093/jac/dkp221
Rasigade, J.-P., Raulin, O., Picaud, J.-C., Tellini, C., Bes, M., Grando, J., et al. (2012). Methicillin-resistant Staphylococcus capitis with reduced Vancomycin susceptibility causes late-onset sepsis in intensive care neonates. PLoS ONE 7:e31548. doi: 10.1371/journal.pone.0031548
Roberts, R. J., Carneiro, M. O., and Schatz, M. C. (2013). The advantages of SMRT sequencing. Genome Biol. 14:405. doi: 10.1186/gb-2013-14-6-405
Shin, J. M., Gwak, J. W., Kamarajan, P., Fenno, J. C., Rickard, A. H., and Kapila, Y. L. (2016). Biomedical applications of nisin. J. Appl. Microbiol. 120, 1449–1465. doi: 10.1111/jam.13033
Siguier, P., Perochon, J., Lestrade, L., Mahillon, J., and Chandler, M. (2006). ISfinder: the reference centre for bacterial insertion sequences. Nucleic Acids Res. 34, D32–D36. doi: 10.1093/nar/gkj014
Stoll, B. J., Hansen, N., Fanaroff, A. A., Wright, L. L., Carlo, W. A., Ehrenkranz, R. A., et al. (2002). Late-onset sepsis in very low birth weight neonates: the experience of the NICHD Neonatal Research Network. Pediatrics 110, 285–291. doi: 10.1542/peds.110.2.285
Stoll, B. J., Hansen, N. I., Higgins, R. D., Fanaroff, A. A., Duara, S., Goldberg, R., et al. (2005). Very low birth weight preterm infants with early onset neonatal sepsis: the predominance of gram-negative infections continues in the National Institute of Child Health and Human Development Neonatal Research Network, 2002-2003. Pediatr. Infect. Dis. J. 24, 635–639. doi: 10.1097/01.inf.0000168749.82105.64
Taft, D. H., Ambalavanan, N., Schibler, K. R., Yu, Z., Newburg, D. S., Deshmukh, H., et al. (2015). Center variation in intestinal Microbiota prior to late-onset sepsis in preterm infants. PLoS ONE 10:e0130604. doi: 10.1371/journal.pone.0130604
Vallenet, D., Belda, E., Calteau, A., Cruveiller, S., Engelen, S., Lajus, A., et al. (2013). MicroScope–an integrated microbial resource for the curation and comparative analysis of genomic and metabolic data. Nucleic Acids Res. 41, D636–D647. doi: 10.1093/nar/gks1194
Vallenet, D., Labarre, L., Rouy, Z., Barbe, V., Bocs, S., Cruveiller, S., et al. (2006). MaGe: a microbial genome annotation system supported by synteny results. Nucleic Acids Res. 34, 53–65. doi: 10.1093/nar/gkj406
Van Der Zwet, W. C., Debets-Ossenkopp, Y. J., Reinders, E., Kapi, M., Savelkoul, P. H., Elburg, R. M., et al. (2002). Nosocomial spread of a Staphylococcus capitis strain with heteroresistance to vancomycin in a neonatal intensive care unit. J. Clin. Microbiol. 40, 2520–2525. doi: 10.1128/JCM.40.7.2520-2525.2002
Venkatesh, M. P., Placencia, F., and Weisman, L. E. (2006). Coagulase-negative staphylococcal infections in the neonate and child: an update. Semin. Pediatr. Infect. Dis. 17, 120–127. doi: 10.1053/j.spid.2006.06.005
Wiedemann, I., Breukink, E., van Kraaij, C., Kuipers, O. P., Bierbaum, G., de Kruijff, B., et al. (2001). Specific binding of nisin to the peptidoglycan precursor lipid II combines pore formation and inhibition of cell wall biosynthesis for potent antibiotic activity. J. Biol. Chem. 276, 1772–1779. doi: 10.1074/jbc.M006770200
Zhang, Y.-Q., Ren, S.-X., Li, H.-L., Wang, Y.-X., Fu, G., Yang, J., et al. (2003). Genome-based analysis of virulence genes in a non-biofilm-forming Staphylococcus epidermidis strain (ATCC 12228). Mol. Microbiol. 49, 1577–1593. doi: 10.1046/j.1365-2958.2003.03671.x
Keywords: bacteremia, multiple drug resistance, late-onset sepsis, SMRT, nisin, comparative genomics, Staphylococcus capitis
Citation: Martins Simões P, Lemriss H, Dumont Y, Lemriss S, Rasigade J-P, Assant-Trouillet S, Ibrahimi A, El Kabbaj S, Butin M and Laurent F (2016) Single-Molecule Sequencing (PacBio) of the Staphylococcus capitis NRCS-A Clone Reveals the Basis of Multidrug Resistance and Adaptation to the Neonatal Intensive Care Unit Environment. Front. Microbiol. 7:1991. doi: 10.3389/fmicb.2016.01991
Received: 07 September 2016; Accepted: 28 November 2016;
Published: 15 December 2016.
Edited by:
Benoit Doublet, National Institute for Agricultural Research (INRA), FranceReviewed by:
Olin Silander, Massey University, New ZealandRavi Ranjan, University of Illinois at Chicago, USA
Copyright © 2016 Martins Simões, Lemriss, Dumont, Lemriss, Rasigade, Assant-Trouillet, Ibrahimi, El Kabbaj, Butin and Laurent. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Patrícia Martins Simões, patricia.martins-simoes01@chu-lyon1.fr