 Heiko Nacke1†*
Heiko Nacke1†* Kezia Goldmann2,3†
Kezia Goldmann2,3† Ingo Schöning4†
Ingo Schöning4† Birgit Pfeiffer1
Birgit Pfeiffer1 Kristin Kaiser1
Kristin Kaiser1 Genis A. Castillo-Villamizar1
Genis A. Castillo-Villamizar1 Marion Schrumpf4François Buscot2,5
Marion Schrumpf4François Buscot2,5 Rolf Daniel1
Rolf Daniel1 Tesfaye Wubet2,5
Tesfaye Wubet2,5- 1Department of Genomic and Applied Microbiology and Göttingen Genomics Laboratory, Institute of Microbiology and Genetics, Georg-August University, Göttingen, Germany
- 2Department of Soil Ecology, UFZ-Helmholtz Centre for Environmental Research, Halle, Germany
- 3Department of Biology II, University of Leipzig, Leipzig, Germany
- 4Max Planck Institute for Biogeochemistry, Jena, Germany
- 5German Centre for Integrative Biodiversity Research (iDiv) Halle-Jena-Leipzig, Leipzig, Germany
The complex interactions between trees and soil microbes in forests as well as their inherent seasonal and spatial variations are poorly understood. In this study, we analyzed the effects of major European tree species (Fagus sylvatica L. and Picea abies (L.) Karst) on soil bacterial and fungal communities. Mineral soil samples were collected from different depths (0–10, 10–20 cm) and at different horizontal distances from beech or spruce trunks (0.5, 1.5, 2.5, 3.5 m) in early summer and autumn. We assessed the composition of soil bacterial and fungal communities based on 16S rRNA gene and ITS DNA sequences. Community composition of bacteria and fungi was most strongly affected by soil pH and tree species. Different ectomycorrhizal fungi (e.g., Tylospora) known to establish mutualistic associations with plant roots showed a tree species preference. Moreover, bacterial and fungal community composition showed spatial and seasonal shifts in soil surrounding beech and spruce. The relative abundance of saprotrophic fungi was higher at a depth of 0–10 vs. 10–20 cm depth. This was presumably a result of changes in nutrient availability, as litter input and organic carbon content decreased with soil depth. Overall bacterial community composition showed strong variations under spruce with increasing distance from the tree trunks, which might be attributed in part to higher fine root biomass near spruce trunks. Furthermore, overall bacterial community composition was strongly affected by season under deciduous trees.
Introduction
Earth currently harbors approximately three trillion trees and only one gram of soil can contain billions of microbial cells (Rosselló-Mora and Amann, 2001; Crowther et al., 2015). The effect of trees on bacteria and fungi in forest soils, comprising many taxa involved in decomposition of plant litter as well as deadwood, is however poorly understood (Wubet et al., 2012; Pfeiffer et al., 2013; Purahong et al., 2014). Forest trees substantially impact soil physical, chemical and biological properties by species-specific stemflow, root architecture, leaf and root litter inputs, root exudates, nutrient uptake, shade, and microclimate (Augusto et al., 2002; Ayres et al., 2009; Raz-Yaseef et al., 2010; Cesarz et al., 2013). As a consequence of direct or indirect tree impacts, changes in the spatial distribution of microbes, vertically through the soil profile as well as horizontally with increasing distance from tree trunks, can occur (Saetre and Bååth, 2000; Ettema and Wardle, 2002). Although numerous studies on the effects of plants on soil microorganisms are available, they rarely focus on microbial communities under trees (Thoms et al., 2010; Urbanová et al., 2015; Uroz et al., 2016). Surveys on effects of pure tree species in a forest stand as well as those focusing on vegetation gradients or chronosequences contributed to the current overall picture concerning tree influences on soil microbial communities (e.g., Cong et al., 2015; Zeng et al., 2016).
European beech (Fagus sylvatica L.) and Norway spruce (Picea abies (L.) Karst) represent dominant forest trees in Central Europe (Cesarz et al., 2013; Hanewinkel et al., 2013). Since the 19th century, reforestation of devastated forest sites using Norway spruce has been very common in Central Europe (Berger and Berger, 2012). Beech forests show a high seasonal variation in aboveground litter input, which is predominately autumnal. In contrast, the aboveground litter input in spruce forest remains relatively constant over the year. Components of needle litter from Norway spruce such as waxes and phenolic compounds are highly recalcitrant to biological degradation, whereas beech leaf litter contains higher amounts of more readily decomposed water-soluble substances (Nykvist, 1963; Priha and Smolander, 1997). Replacement of beech by spruce species is therefore accompanied by changes in humus form, acidity and soil structure (Berger and Berger, 2012). Upper soil horizons are dominated by leaf litter input, and roots; their residues and exudation patterns shape the subsoil (Moll et al., 2015). Spruce is typically shallow-rooted, whereas beech has a deep rooting system (so called “base-pump”). Consequently, variation in nutrient availability affects microbial communities along soil depths (Huang et al., 2013; Moll et al., 2015). Between Fagus sylvatica L. and Picea abies (L.) Karst, the quantity and composition of exudates varies with season (Geßler et al., 1998; Fender et al., 2013) and potentially affects microbial processes such as respiration (Cesarz et al., 2013).
European beech and Norway spruce forest stands differ in the magnitude of stemflow. In beech stands, stemflow water contributes 5–20% to the annual soil water input (Koch and Matzner, 1993; Johnson and Lehmann, 2006). Stemflow in conifer forests is often below 1% due to differences in branch angle, specific surface roughness of branches and bark (Johnson and Lehmann, 2006). The high stemflow in beech forests is associated by a decrease of soil pH next to the stem base versus the surrounding soil (Koch and Matzner, 1993). A similar effect has not been demonstrated in Norway spruce forest.
Previous studies have largely used methods providing coarse phylogenetic information to identify effects of forests on soil microbial communities. Using automated ribosomal intergenic spacer analysis (ARISA), ester linked fatty acid methyl ester (EL-FAME) analyses, and denaturing gradient gel electrophoresis (DGGE), differences in soil bacterial and fungal community structure in temperate broad-leaved and coniferous forests have been reported (Lejon et al., 2005; Zechmeister-Boltenstern et al., 2011; Jiang et al., 2012). Recently, Tedersoo et al. (2016) analyzed pyrosequencing-derived ITS sequences to assess the effects of tree diversity on fungi, protists and meiofauna inhabiting forest soil. Results indicated that compared to the effects of individual tree species and soil parameters, tree diversity per se had a minor influence on the taxonomic richness of soil biota (Tedersoo et al., 2016). In addition, based on amplicon pyrosequencing data, significant effects of tree species on soil bacterial and fungal community composition were reported by Urbanová et al. (2015).
While several recent marker gene sequencing-based studies focused either on bacteria or fungi in forest soils, they have rarely been considered together (Yarwood et al., 2010; Baldrian et al., 2012; Urbanová et al., 2015). Fungi are typically larger in size than bacteria and exhibit a higher biomass. Therefore, they interact with their environment, e.g., by moving water and nutrients, on a larger spatial scale compared to bacteria (Coleman and Crossley, 1996; van der Heijden et al., 2008; Trevors, 2010), which might result in a more homogeneous distribution of fungal communities in soil. The life cycle of both bacteria and fungi inhabiting forest soils can be strongly affected by seasons through changes in abiotic and biotic factors (Thoms and Gleixner, 2013).
In this study, we applied pyrosequencing of the V3–V5 region of the 16S rRNA gene and the ITS DNA region to assess composition of soil bacterial and fungal communities in a European beech and a Norway spruce forest. We considered potential seasonal variation in microbial communities by collecting samples in early summer and autumn. Furthermore, to determine spatial tree effects, soil collected from different depths and horizontal distances toward tree trunks was considered within this survey. We examined the following hypotheses: (1) bacterial and fungal community composition are affected by tree species, (2) the relative abundance of saprotrophic microorganisms decreases with soil depth, (3) bacteria respond stronger to growing distance from trees than fungi, and (4) seasonal variation of soil bacterial and fungal community composition is stronger under deciduous versus coniferous forests.
Materials and Methods
Sites and Soil Sampling
All soil samples were derived from a beech (Fagus sylvatica L.) and a spruce (Picea abies L. (Karst)) forest site (distance between the two forest sites: approximately 5 km) located in the Hainich-Dün region in Germany (Fischer et al., 2010). The beech and spruce forest stands were originally established as plantations and are managed (management type, age class forest) since 1760 and approximately 1930, respectively (Wäldchen et al., 2011). Due to the very fertile soils (the original parent material was limestone covered by loess) at both sites, beech-dominated forest would be the natural forest type. The age of the trees at both sites ranged between 50 and 65 y. Beech and spruce trees exhibited average crown radii of 387 ± 29 and 209 ± 12 cm, respectively. The mineral soil was sampled at 0–10 cm and 10–20 cm depth using a split tube sampler with a diameter of 4.8 cm (Eijkelkamp Agrisearch Equipment, Giesbeck, Netherlands). Mineral soil samples were taken from different horizontal distances (0.5, 1.5, 2.5, and 3.5 m) from the trunks of four randomly-selected trees per site (“tree distance”; see Figure 1). Sampling was performed in two seasons, early summer and autumn 2012. Five year averages (2008–2012) of soil temperature, measured at a depth of 10 cm, showed similar seasonal variations in the beech (May: 12.1°C, November: 4.1°C) and spruce forest stand (May: 12.7°C, November: 4.2°C). We applied a paired sampling. The sampling positions in autumn were <30 cm away from the sampling points in early summer (Table S1). All sampling points showed a distance >3.5 m to tree trunks (except trunks of the four selected beech and spruce trees, respectively). In total 128 soil samples (2 sites × 2 seasons × 4 replicate trees × 4 horizontal distances × 2 soil depths) were immediately sieved to <4 mm in the field and individually homogenized. One subsample (>200 g) of each sample was air-dried and sieved to <2 mm for soil chemical analyses and another subsample (50 g) was frozen (−20°C) for extraction of nucleic acids.
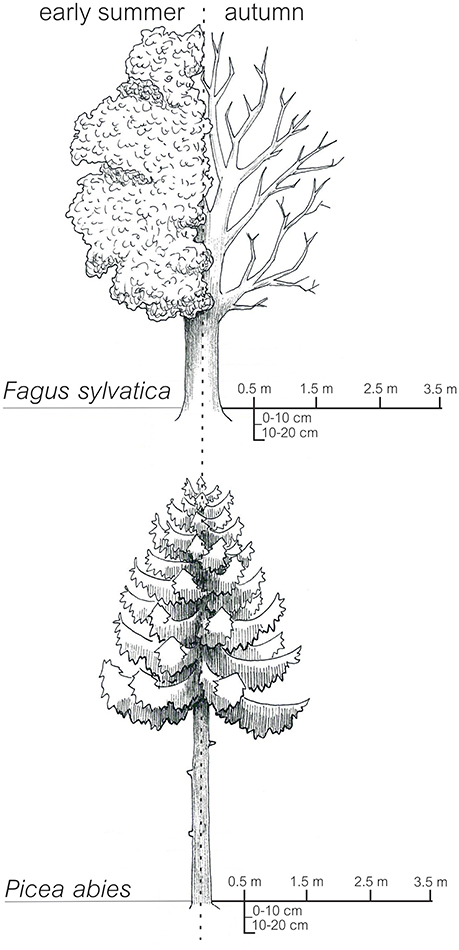
Figure 1. Sampling design: In early summer and autumn 2012 samples were taken at a distance of 0.5, 1.5, 2.5, and 3.5 m from the tree trunks of four European beech and four Norway spruce trees. At all sampling points soil samples from 0–10 cm and 10–20 cm were taken.
Soil Physical and Chemical Properties
Soil pH was measured in duplicate in the supernatant of 1:2.5 mixtures of soil and aqueous 0.01 M CaCl2 with a glass electrode. Additionally, the gravimetric water content of the air-dried soil was determined. The empirical equation of Wäldchen et al. (2012) was used to estimate clay content in the samples. The remaining soil was ground to <100 μm. Ground samples were analyzed for total carbon (TC) and nitrogen (TN) by dry combustion with the CN analyzer “Vario Max”™ (Elementar Analysensysteme GmbH, Hanau, Germany). Inorganic carbon (IC) concentrations were determined with the same analyzer after the ignition of samples for 16 h at 450°C. The organic carbon (OC) concentrations equaled the differences between TC and IC.
DNA Extraction, Amplification and Pyrosequencing
Total microbial community DNA was extracted from approximately 2 g of frozen soil per sample using the PowerSoil™ total RNA isolation kit, the PowerSoil™ DNA elution accessory kit, and the PowerClean™ DNA Clean-Up kit (MoBio Laboratories, Carlsbad, CA, USA) according to the instruction. DNA concentrations were quantified using a NanoDrop UV-Vis spectrophotometer (Peqlab Biotechnologie GmbH, Erlangen, Germany).
The V3–V5 region of bacterial 16S rRNA genes was amplified by PCR. The following set of primers containing Roche 454 pyrosequencing adaptors (underlined) and a sample-specific MID (Extended Multiplex Identifier) was used: V3for 5′-CCATCTCATCCCTGCGTGTCTCCGACTCAG-MID-TACGGRAGGCAGCAG-3′ (Liu et al., 2007) and V5rev 5′-CCTATCCCCTGTGTGCCTTGGCAGTCTCAGCCGTCAATTCMTTTGAGT-3′ (Wang and Qian, 2009). The PCR reaction mixture (50 μl) contained 10 μl 5-fold reaction buffer (Phusion HF buffer, Thermo Fisher Scientific Inc., Germany), 200 μM of each of the four deoxynucleoside triphosphates, 5% DMSO, 1 U Phusion high fidelity DNA polymerase (Thermo Fisher Scientific Inc.), approximately 25 ng DNA as template, and 4 μM of each of the primers. The PCR reactions were initiated at 98°C (2 min), followed by 25 cycles of 98°C (45 s), 58°C (45 s), and 72°C (40 s), and ended with incubation at 72°C for 5 min.
Fungal ITS DNA was amplified using primer ITS1F (Gardes and Bruns, 1993) containing a sample-specific MID and Roche 454 pyrosequencing adaptor B and primer ITS4 (White et al., 1990) containing Roche 454 pyrosequencing adaptor A. The PCR reactions were performed in a total volume of 50 μl reaction mix containing 1 μl DNA template (7–15 ng), 25 μl Go Taq Green Master mix (Promega, Mannheim, Germany) and 1 μl 25 pmol of each of the ITS region-specific primers. Touchdown PCR conditions as described by Wubet et al. (2012) were used to amplify fungal ITS DNA.
All samples were amplified in triplicate, purified using the peqGold gel extraction kit (Peqlab Biotechnologie GmbH) and the Qiagen gel extraction kit (Qiagen, Hilden, Germany) as recommended by the manufacturer, and pooled in equal amounts. Quantification of PCR products was performed using the Quant-iT dsDNA BR assay kit and a Qubit fluorometer (Life Technologies GmbH, Karlsruhe, Germany). Sequences of partial 16S rRNA genes and fungal ITS DNA were decoded at the Göttingen Genomics Laboratory and the Department of Soil Ecology (UFZ-Helmholtz Centre for Environmental Research, Halle, Germany), respectively, using a Roche GS-FLX 454 pyrosequencer (Roche, Mannheim, Germany) and Titanium chemistry as recommended by the manufacturer.
The 16S rRNA gene and ITS DNA sequences were deposited in the National Center for Biotechnology Information (NCBI) Sequence Read Archive (SRA) under study accession numbers SRP040766 and SRP044665, respectively.
Sequence Analysis
Bacterial 16S rRNA gene sequence datasets were preprocessed as described by Broszat et al. (2014). Briefly, bacterial sequences shorter than 200 bp, as well as those exhibiting low quality values (<25), more than two primer mismatches, or long homopolymers (>8 bp), were removed using QIIME (Caporaso et al., 2010). In addition, the bioinformatics tools cutadapt (Martin, 2011), Uchime (Edgar et al., 2011), and Acacia (Bragg et al., 2012) were used for truncation of remaining primer sequences, removal of potential chimeric sequences, and removal of noise introduced by amplicon pyrosequencing. Uclust (Edgar, 2010), implemented in QIIME (Caporaso et al., 2010), was used to determine bacterial OTUs at a genetic distance of 3%. To taxonomically classify OTUs, partial 16S rRNA gene sequences were compared with the SILVA SSU database release 119 (Pruesse et al., 2007). OTUs classified as chloroplast or mitochondrion and unclassified OTUs (proportion of unclassified OTUs was approximately 0.2%), which were not affiliated to bacteria, were removed from 16S rRNA gene sequence datasets.
Fungal ITS DNA sequence datasets were preprocessed with Mothur (Schloss et al., 2009) as described by Goldmann et al. (2015). In brief, sequences with ambiguous bases, homopolymers and primer differences (>8 bp) as well as MIDs were removed in a first filtering step. Simultanously, short reads (<300 bp), sequences with a low quality score (<20) and noisy sequence ends were removed. Samples were checked for chimeric sequences using the UCHIME algorithm (Edgar et al., 2011). Cd-hit (Li and Godzik, 2006) was applied to determine fungal OTUs at 3% genetic distance. To identify fungi and taxonomically classify OTUs, ITS DNA sequences were queried against the UNITE database (Kõljalg et al., 2013) by using the classify.seq command as implemented in MOTHUR (Schloss et al., 2009). All produced OTUs belonged to the kingdom fungi. To improve the taxonomical resolution, OTUs that had been assigned only down to the family level were subjected to a BLASTn search (e.g., Johnson et al., 2008) against the NCBI GenBank database (Benson et al., 2015). The searches excluded uncultured and environmental sample sequences and only assignments with a query cover >95%, E <0.0001 and sequence identity >97% were considered. Finally, all fungal OTUs identified at the genus level were grouped into ectomycorrhizal, saprotrophic, and other fungi based on literature.
Bacterial and fungal OTUs comprising only one or two sequences (singleton and doubleton OTUs) were removed from the datasets. The number of analyzed sequences per sample can have an effect on the predicted number of OTUs (Morales et al., 2009). Therefore, OTU-based comparisons were performed at the same level of surveying effort (bacteria: 2540 sequences per sample; fungi: 1996 sequences per sample). In this study, we focused on microbial community composition. Data on microbial diversity is provided in the Supplementary Material (see Figures S1, S2). OTUs identified at a genetic distance of 3% were used to calculate rarefaction curves and the Shannon index.
Statistical Analyses
The response of main soil characteristics (e.g., C:N ratio, clay content) to soil depth (0–10 and 10–20 cm depth), season (early summer and autumn) and tree distance (0.5, 1.5, 2.5, and 3.5 m) was assessed for both study sites separately by analysis of covariance (ANCOVA) using the “aov” command of the “Stats” R-package (R Development Core Team, 2015). The random effects of the four sampling transects per study site were considered in the analysis by including them as a factor in our linear models (tree replicate).
The effect of tree species on soil bacterial and fungal community composition, respectively, was visualized using principal coordinates analysis plots generated with the emperor software package (Vázquez-Baeza et al., 2013) and the “ordiplot” function incorporating environmental vectors calculated with the “envfit” function of the “Vegan” R-package (Oksanen et al., 2016). In order to test the effects of tree replicate, soil pH, OC, soil depth, sampling season, and tree distance on bacterial and fungal community composition, we performed multivariate analysis of variance (MANOVA) using the “adonis" command of the “Vegan” R-package (Oksanen et al., 2016) based on weighted UniFrac (Lozupone et al., 2011) distance matrices. The adonis function in R implements a sequential sum of squares (type 1). A priori we decided to include first the random variance of the tree replicates and important abiotic drivers (soil pH and organic C) into the model. In a second step the factors depth, season and distance were added. This means that the significance of depth, season and distance was examined after removal of variance explained by soil pH and organic C concentration. Changing the order of soil pH and organic C or the order of depth, season and distance in the model would not change the significance of the individual factors. This can be explained by the missing collinearity among these factors. These analyses were conducted for whole microbial communities and microbial communities under each tree species individually. Adjusted R2-values of total models increased with the addition of every single considered parameter (Tables S2, S3).
To further identify individual taxa strongly associated with a specific tree species, season or spatial position in soil, the multipatt algorithm and the “IndVal” function in the “Indicspecies” R-package (De Cáceres and Legendre, 2009) was used based on bacterial and fungal OTUs. The PAST statistical package (Hammer et al., 2001) was used for the performance of Mann-Whitney U test and Spearman's rank correlations. We applied Mann-Whitney U test to identify dominant genera showing significant differences between sets of samples. Spearman's rank correlations were used to correlate relative abundances of dominant genera with soil parameters.
Results
General Characteristics of Soil Samples
Both forest stands grow on limestone, which is covered with a loess layer of variable thickness. The loess layer is thinner at the spruce than at the beech forest site. Therefore, in 0–10 cm depth pH values ranged between 3.1 and 5.9 at the spruce site and between 3.7 and 4.4 at the beech site (Table 1, Table S4). The pH values determined for our samples are typical for the two forest sites. At 5 out of 32 sampling locations within the spruce forest the pH at a depth of 0–10 cm was >5.5 indicating that the loess layer was less pronounced or absent and that the parent material mainly originated from limestone. We did not detect a decrease of the soil pH next to the stem basis of beech trees (0.5 m tree distance) compared to the other considered sampling distances (Table S4). At a depth of 10–20 cm the average pH increased by 0.9 units in the spruce stand, whereas it decreased by 0.2 units in the beech stand, which is again a result of the lower loess layer thickness in the spruce compared to the beech stand. This was confirmed by the clay content (0–10 cm), which was with 388 ± 15.2 g kg−1 (mean ± standard error) on average higher at the spruce than at the beech site with 276 ± 4.4 g kg−1. At the 0–10 cm depth, the soils contained on average 32.6 ± 2.3 g kg−1 and 26.2 ± 0.8 g kg−1 OC in the spruce and beech stand. The OC concentrations decreased with depth. Organic C concentrations at the 0- to 10-cm depth were strongly related to estimated clay contents (r = 0.79, P < 0.001). Due to collinearity between OC concentration, clay content, and C:N ratio, we only included OC concentration in subsequent statistical analyses.

Table 1. Basic properties of soil samples derived from the beech and spruce stands.
Soil Bacterial and Fungal Community Profiles
Pyrotag processing yielded a total of 864,096 bacterial and 255,488 fungal high-quality sequences with an average length of 464 and 300 bp, respectively. At a genetic distance of 3%, 23,727 bacterial and 1336 fungal OTUs were identified across all analyzed soil samples. In the final microbial dataset, the number of OTUs per individual soil sample ranged from 505 to 1440 (bacteria) and 45 to 191 (fungi). Taxonomic classification was based on closest matches of OTUs to particular phylogenetic groups. Each of the dominant phyla and genera identified in this study (see Figures 2, 3) is represented by more than one OTU determined at a genetic distance of 3%. The bacterial phyla and proteobacterial classes detected in each of the individual soil samples comprised Acidobacteria (average relative abundance: 40.7 ± 0.8%), Alphaproteobacteria (20.5 ± 0.4%), Actinobacteria (9.4 ± 0.3%), Gammaproteobacteria (5.8 ± 0.2%), Chloroflexi, (4.8 ± 0.2%), Gemmatimonadetes (4.4 ± 0.2%), Deltaproteobacteria (3.8 ± 0.2%), Betaproteobacteria (3.3 ± 0.1%), Bacteroidetes (2.1 ± 0.1%) and candidate division WPS-2 (1.5 ± 0.1%) (Figure 2). Genus level analysis of the bacterial community showed high relative abundances (average relative abundance of each genus >1%) of Bradyrhizobium followed by Acidothermus, Gemmatimonas, Rhizomicrobium, and Reyranella (Figure 3). Acidobacteria represent the most abundant phylum in our study. Subgroup 2 (average relative abundance: 14.1 ± 0.6%), subgroup 1 (11.1 ± 0.5%), subgroup 3 (10.1 ± 0.3%), and subgroup 6 (2.8% ± 0.3%) showed the highest average relative abundance among acidobacterial representatives.
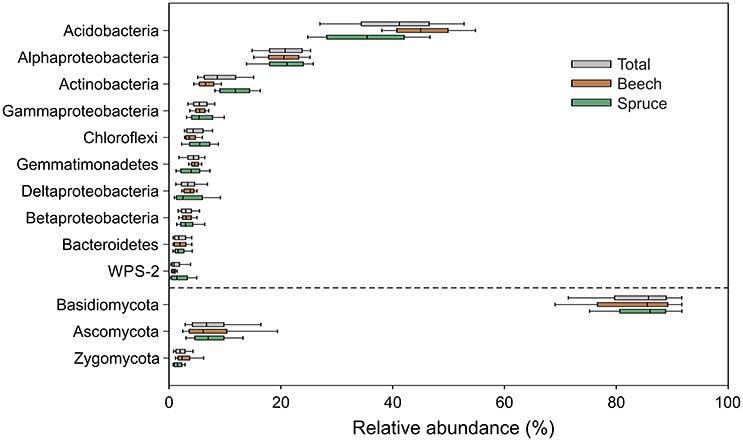
Figure 2. Box-and-whiskers plot showing relative abundances of bacterial and fungal phyla as well as proteobacterial classes detected in each of the analyzed 128 soil samples. Relative abundances of taxa across all samples (gray color) as well as separately with respect to soil surrounding beech (brown color) and spruce (green color) are depicted. The dashed line separates relative abundances of bacterial and fungal taxa.
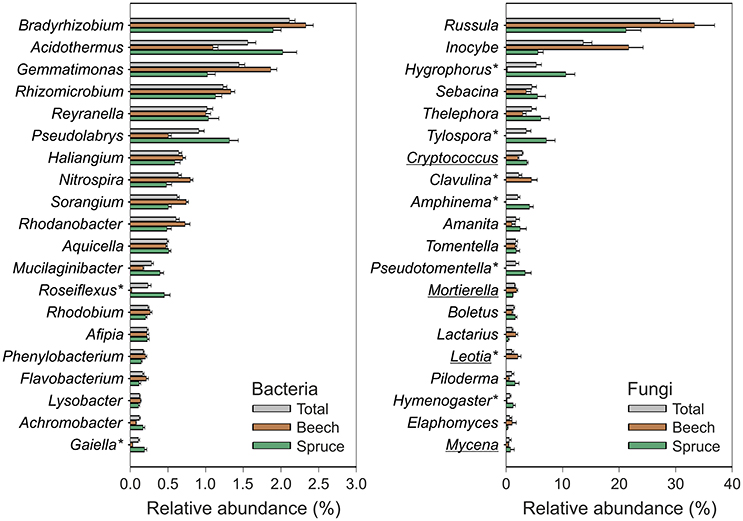
Figure 3. Relative abundance of dominant bacterial and fungal genera detected in the analyzed soil samples. The data represent mean values and standard errors of relative abundance for the 20 most abundant bacterial and fungal genera, respectively. Acidobacteria were analyzed at the subgroup level and therefore not considered within this figure. Relative abundances of taxa across all samples (gray color) as well as separately with respect to soil surrounding beech (brown color) and spruce (green color) are depicted. Asterisks indicate taxa showing an at least five-fold difference in mean relative abundance between spruce and beech (P < 0.001 for the Mann-Whitney U test). Underlined taxa: saprotrophic fungi (all other depicted fungal genera represent ectomycorrhizal fungi).
The fungal community was dominated by Basidiomycota (average relative abundance: 87.7 ± 0.7%), followed by Ascomycota (8.9 ± 0.6%), and Zygomycota (2.5 ± 0.2%) (Figure 2). In total, 89% of all dominant fungal OTUs were assigned to more than 200 fungal genera. The most abundant fungal genera were Russula (average relative abundance: 33.3 ± 2.7%), followed by Inocybe (16.8 ± 1.8%), Hygrophorus (6.2 ± 1.0%), Sebacina (5.7 ± 1.0%), and Thelephora (5.6 ± 1.0%) (Figure 3). Functional group assignment of the fungal communities revealed that among the 20 most abundant fungal genera (Figure 3), 16 are known to be ectomycorrhizal (ECM) fungi, whereas the remaining four have a saprotrophic lifestyle (Cryptococcus, Mortierella, Leotia, and Mycena).
Tree Species Effects on Microbial Community Composition
Samples collected under beech and spruce tend to cluster separately in principal coordinates analysis plots (Figure 4). The axes of these plots explain less of the variability in fungal community composition (axis 1 = 14%) compared to bacterial community composition (axis 1 = 41%). The variation explained by tree species was 13.8% (P < 0.001) in bacterial and 14.9% (P < 0.001) in fungal communities (Table S2). Furthermore, tree species (European beech or Norway spruce) had a stronger impact on soil bacterial and fungal community composition than soil depth, distance from tree trunk or season (Table S2). We identified specific indicator OTUs for soils surrounding beech or spruce stands (Table S5). Each bacterial indicator OTU showed an average relative abundance <1%, whereas few fungal indicator OTUs showed relative abundances >1%. Detailed information on relative abundances for all indicator OTUs is provided in Table S5.
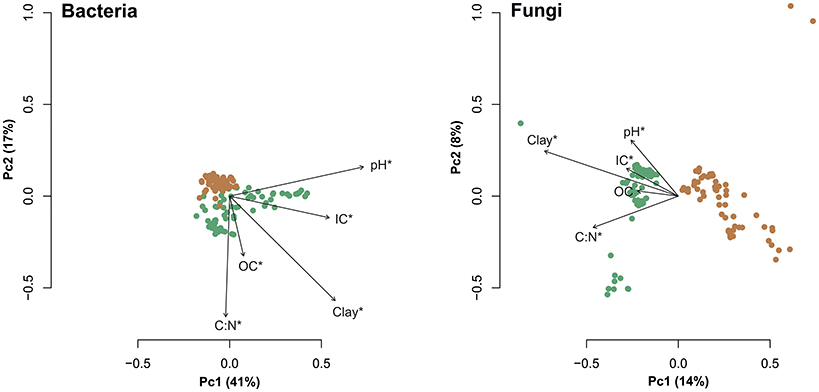
Figure 4. Principal coordinates analysis plots based on weighted UniFrac distances calculated at 3% genetic distance. Brown circles represent samples derived from beech surrounding soil and samples derived from spruce surrounding soil are depicted as green circles. Vectors represent response variables pH, estimated clay content, C:N ratio, organic carbon (OC), and inorganic carbon (IC). Significant values (P < 0.05) according to “envfit” calculations are indicated by asterisks.
For bacteria, 13 indicator OTUs were determined at the beech site and 10 indicator OTUs at the spruce site. The majority of bacterial OTUs representing indicators at the beech site were affiliated to Acidobacteria (mainly subgroup 2) (Table S5). Indicators at the spruce site comprised Chloroflexi, WD272 and several Acidobacteria subgroup 1 OTUs.
For both tree species, eight fungal OTUs were identified as potential indicators (Table S5). Under beech, a saprotrophic Mortierella elongata OTU and a Trichoderma OTU and ECM fungi OTUs (a Russula cyanoxantha OTU and a Xerocomus chrysenteron OTU) were identified as indicator OTUs. Indicators for spruce were three OTUs classified as saprotrophic fungi (Exophiala and two Penicillium OTUs). The two indicator ECM fungi under spruce were Hygrophorus and Amphinema.
Microbial community composition under both tree species was significantly affected by tree replicate, soil pH and OC (Table 2). Among the analyzed factors soil pH and tree species explained most of the variation in microbial community composition (Table S2).
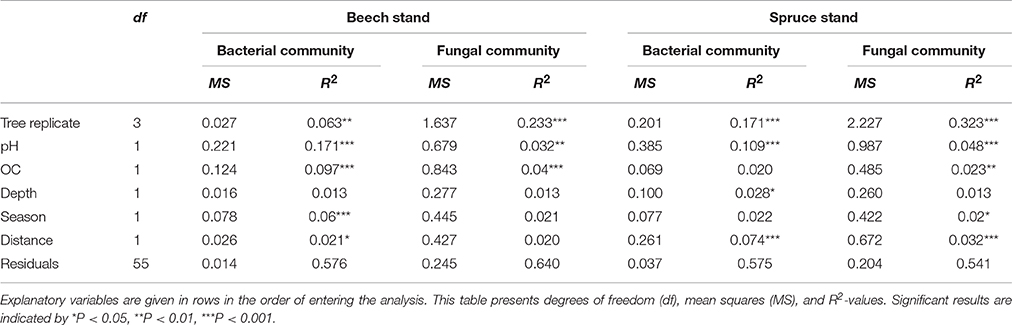
Table 2. Multivariate analysis of variance based on weighted UniFrac distances with tree replicate, pH, OC, soil depth, season and distance as response variable.
Spatial and Seasonal Variability of Soil Microbial Community Composition
Bacterial community composition varied significantly with depth under spruce (Table 2). We found that relative abundance of OTUs of the dominant genus Gaiella was negatively correlated with OC concentration (P < 0.001) and higher at the 10- to 20-cm depth than the 0- to 10-cm depth. The relative abundance of the bacterial genus Mucilaginibacter also showed variations with soil depth. It was higher at the 0- to 10-cm depth vs. the 10- to 20-cm depth (P < 0.001). The fungal community composition showed no correlation with soil depth under both tree species (Table 2). However, the detected saprotrophic fungi were associated with the upper (0–10 cm depth) mineral soil layers, which were rich in OC (Figure 5). Additionally, the indicator species analysis identified mainly saprotrophic OTUs in the upper 10 cm of the studied soils (Table S5).
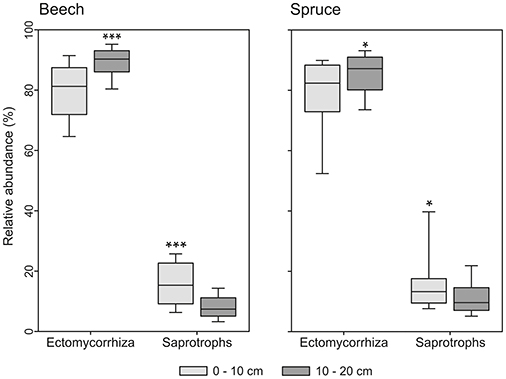
Figure 5. Box-and-whiskers plots showing relative abundance of ectomycorrhizal and saprotrophic fungi under beech and spruce in relation to soil depths. The asterisks indicate significant differences between soil depths for each ecological group determined by ANOVA; *significant (P < 0.05), ***highly significant (P < 0.001).
Spatial horizontal variation of overall bacterial community composition was significant in soil under beech (P < 0.05) and under spruce (P < 0.001) (Table 2). We found that relative abundance of the dominant bacterial genus Nitrospira was significantly higher at 3.5 m vs. 0.5 m distance from spruce trees. Furthermore, a Nitrospirales OTU was identified as an indicator for tree distances of 2.5 and 3.5 m (Table S5). Under beech trees, the relative abundance of Pseudolabrys differed significantly between 0.5 m and 3.5 m horizontal tree distance. Higher relative abundance was detected in soil located close to tree trunks. This effect was recorded with respect to both analyzed soil depths (P < 0.05). Overall fungal community composition differed significantly at different horizontal tree distances only in soil of the spruce stand (P < 0.01) (Table 2). However, fungal indicator species for certain combinations of tree distances were found in beech (Table S5) and spruce stands (Table S5).
A significant seasonal effect on bacterial community composition was detected in soil under beech (P < 0.001) (Table 2). Sequences corresponding to the Rhizobiales (Bradyrhizobium and Rhodobium) showed significantly higher relative abundance in autumn versus early summer (P < 0.001). Consistently, the analysis of indicator species identified an OTU affiliated to Bradyrhizobium in soil under beech in autumn (Table S5). A seasonal impact on fungal community composition was found in soil of the spruce stand (P < 0.05) (Table 2). Two fungal indicator species were identified in early summer in the spruce stand (Table S5). Fungal indicator species for both seasons (autumn and early summer) occurred under beech (Table S5).
Discussion
Selective Association of Tree Species, Bacteria, and Fungi
Differences in distribution of microbial taxa were identified between soil under beech and spruce. This was expected, as even tree genotype within a species can have significant impacts on microbial communities (Schweitzer et al., 2008). A Chloroflexi OTU was identified as indicator for soil surrounding spruce. As several potential genes involved in phytochemical breakdown have been identified in Chloroflexi (Hug et al., 2013; Houghton et al., 2015), it is possible that this indicator microorganism plays a role in decomposition of spruce litter. Furthermore, the occurrence of several members of Acidobacteria was significantly affected by tree species. It can be assumed that acidobacterial taxa contribute to decomposition in forest soils, as genomic and culture characteristics of subgroup 1 and 3 strains have been shown to utilize plant-derived biopolymers (Ward et al., 2009; García-Fraile et al., 2015). Shifts in occurrence of acidobacterial representatives between soil under European beech and Norway spruce might imply preferences for leaf or needle litter. A study on composition of bacterial communities under different deciduous and coniferous trees (e.g., Picea and Fagales species) in Czech forest stands also indicated litter preferences of Acidobacteria (Urbanová et al., 2015).
Forest vegetation (in particular dominant tree species) is important for distribution of mutualistic and saprotrophic fungi (Lauber et al., 2008; Goldmann et al., 2015). ECM fungi (e.g., Russula, Inocybe, Piloderma) establish mutualistic associations with plant roots (Smith and Read, 2008) and show preferences for particular tree species (Ishida et al., 2007; Thoms et al., 2010). In accordance with our study, Goldmann et al. (2015) and Miyamoto et al. (2015) reported a preference of Tylospora for coniferous trees. Some identified fungal indicators under beech (Mortierella elongata, Trichoderma, Russula cyanoxantha) are known to be widespread not just under a certain tree species (Wuczkowski et al., 2003; Grebenc and Kraigher, 2007; Nagy et al., 2011). In contrast, the ECM fungus Xerocomus chrysenteron is known to have a preference for beech (Shi et al., 2002). Indicator species for spruce included three OTUs classified as saprotrophic fungi. Exophiala has already been described as a fungal genus decaying leafs in rainforests (Polishook et al., 1996) or existing as rhizospheric associates in temperate sites (Summerbell, 2005). Another two Penicillium OTUs were identified as saprotrophic indicators for spruce. Previous research (Johansson and Marklund, 1980) reported Penicillium to be antagonistic to Fomes, a well-known fungus infecting spruce trees (Schmidt, 2013). The indicative ECM fungi under spruce, Hygrophorus and Amphinema, were abundant and previously described for spruce ecosystems (Scattolin et al., 2008; Velmala et al., 2013).
Under both tree species, microbial community composition was significantly affected by pH and OC concentration. Noteworthy, among the analyzed factors soil pH and tree species explained most of the variation in overall community composition of bacteria and fungi. Several previous studies have identified soil pH as a major driver of soil bacterial community composition across different regions and land use types (e.g., Lauber et al., 2009; Nacke et al., 2011). In accordance with our results, pH also explained a substantial fraction of variance in microbial community composition within other deciduous and coniferous forest soils (Lauber et al., 2009; Thoms et al., 2010; Goldmann et al., 2015). Furthermore, experiments including addition of substrates such as cellulose, lignin, and glucose to soil showed that the quantity of OC can have a significant impact on soil microbial community composition (Nakatsu et al., 2005; Goldfarb et al., 2011).
Relative Abundance of Saprotrophic Fungi Decreases with Soil Depth
Previous surveys based on DGGE analysis as well as Sanger sequencing and pyrosequencing of 16S rRNA genes have revealed differences in bacterial community composition between topsoil and subsoil (Hansel et al., 2008; Eilers et al., 2012; Huang et al., 2013). This is a result of changes in soil characteristics such as organic C or N concentrations along soil profiles (Hansel et al., 2008; Will et al., 2010). Consistently, relative abundances of the bacterial genus Gaiella, which were higher in 10–20 cm depth than in 0–10 cm depth, were negatively correlated with organic C concentration. Different Mucilaginibacter representatives are capable of pectin, xylan, and laminarin degradation (Pankratov et al., 2007). Mucilaginibacter was more abundant in topsoils (0–10 cm). The genus has been previously associated with cellulose decomposition based on stable isotope probing (Štursová et al., 2012). Leaf and needle litter contains high amounts of the plant cell wall components xylan, pectin, and cellulose, and enters the upper mineral soil first, perhaps explaining the distribution of Mucilaginibacter OTUs.
Recently, McGuire et al. (2013) found discrete fungal communities in different soil horizons in boreal and tropical forest. This can be explained by changing carbon and nutrient contents in soil combined with fungal enzymatic decay abilities (McGuire et al., 2010; Prescott, 2010). Our results (Table 2) showed that fungal taxa in temperate forests do not underlay similar mechanisms as found previously. However, we identified different saprotrophic fungi showing preferences for the upper (0–10 cm depth) mineral soil layer, which was rich in OC. Influenced by the litter layer, the upper 10 cm show high habitat heterogeneity, competition amongst fungi for space, carbon and other soil nutrients (Kadowaki et al., 2014). ECM fungal taxa receive carbon through mycelium connected to plant roots (Smith and Read, 2008). In this study, ECM fungi were abundant irrespective of soil depth since these fungi are not C-limited and may colonize deeper soil layers (McGuire et al., 2013).
Bacteria Are Affected by horizontal Tree Distance under Beech and Spruce
Soil microbial community composition showed higher variability with respect to tree distance under spruce trees versus beech. It is known that spatial distribution of soil microbes can reflect the zone of influence and positioning of individual trees in forests (Saetre and Bååth, 2000; Ettema and Wardle, 2002). As stemflow was shown to significantly decrease soil pH, specifically close to beech trees (Koch and Matzner, 1993), we expected a clear change in microbial community composition next to beech trunks (0.5 m tree distance). However, we could neither detect a decrease in pH at 0.5 m distance to beech trunks, nor a strong change in microbial community composition next to the beech trees. Spatial horizontal variations in bacterial community composition under beech and spruce, recorded in this study, might have been partly evoked by changes in root activities with respect to varying tree distances. N demand of spruce trees in summer and autumn is mainly met by uptake of N compounds from soil and subsequent transport of reduced N from the roots to the shoot via the transpiration stream (Weber et al., 1998). Due to a negative relationship between fine root biomass and tree distance (steep decrease of fine root biomass at tree distances >2 m) (Petritan et al., 2011), uptake of N compounds via roots might be more pronounced in soil located close to the analyzed coniferous tree trunks. This potentially explains the spatial horizontal variations in occurrence of nitrifying bacteria belonging to Nitrospirales under spruce.
Under beech, relative abundance of Pseudolabrys was significantly affected by horizontal tree distance. Only one Pseudolabrys species, isolated from Taiwanese soil, has been described (Kämpfer et al., 2006). In our study, more than one OTU determined at a genetic distance of 3% was affiliated to Pseudolabrys. The taxon Pseudolabrys, representing one of the most abundant genera detected in this study, belongs to the Rhizobiales, which are known to interact with plants (Erlacher et al., 2015). Changes in root densities or activities may be a major reason for high relative abundance of Pseudolabrys in soil located close to beech trunks.
Branco et al. (2013) found that an increase in soil pH with pine tree distance was related to changing occurrence of fungal species. Variation in pH at different tree distances (Table S4) also account for changes in fungal community composition under the conifer trees analyzed in our study (P < 0.05) (Table 2).
More Seasonal Soil Community Variation in Beech than in Spruce Forests
Soil bacterial community composition under beech was strongly affected by season (P < 0.001). Recently, López-Mondéjar et al. (2015) reported that bacterial communities undergo seasonal changes in mineral soil of a Quercus petraea (Matt.) Liebl forest. They assume that seasonal differences in the activity of tree roots are a major driver of soil bacterial community composition in deciduous forest. Here, we found that different members of the Rhizobiales were more abundant under beech in autumn than in early summer. As Rhizobiales are known to interact with plants, seasonal root impacts might affect their abundance in temperate deciduous forest. Understory vegetation varies between European beech and Norway spruce age class forests in the study region (Boch et al., 2013). It is possible that the Rhizobiales community is affected by seasonal changes in understory vegetation. Furthermore, seasonal shifts in soil moisture and temperature may also affect bacterial community composition in the analyzed soil (Kaiser et al., 2010; Shay et al., 2015).
Seasonal impacts on fungi were reported previously (e.g., Stevenson et al., 2014; Moll et al., 2015). In this study, soil fungal community composition was affected by season under spruce (P < 0.05) but not as expected under beech. Recently, Voříšková et al. (2014) also detected no significant seasonal effect on fungal community composition in soil of a deciduous forest (oak forest near Prague, Czech Republic). Nevertheless, in the litter horizon, which was not analyzed in our study, seasonal changes in fungal community composition were identified by Voříšková et al. (2014). These changes are associated with nutrient input from fresh litter, which occurs in temperate deciduous forests each autumn (Voříšková et al., 2014). In accordance with our study, Lin et al. (2016) reported seasonal shifts of fungi in coniferous forests. The air and soil temperatures at both forest stands were higher in early summer, whereas the soil water content was increased in autumn (Table S6). Hence, comparable weather conditions would suggest similar fungal reactions toward changing season at the beech and spruce stand. However, a relatively thick needle litter layer (~8 cm) was removed before soil sampling under spruce. Breakdown of needles, which are highly recalcitrant to biological degradation, is mainly performed by fungi. It is possible that the distinct fungi colonizing needles (Korkama-Rajala et al., 2008) and consequently soil fungal communities under coniferous trees are susceptible to climatic changes in autumn. In addition, unmeasured factors might account for the shifts of fungal communities under spruce. Future studies can evaluate if these findings are artificial or ecologically reasonable.
Conclusion
In accordance with our first hypothesis, beech and spruce trees strongly shaped the community composition of soil bacteria and fungi in temperate forests. Tree species-specific preferences with respect to bacterial and fungal microorganisms, such as a Chloroflexi representative, members of Acidobacteria subgroup 2 or Hygrophorus and Clavulina, were identified. Trees also have manifold impacts on the seasonal and spatial distribution of soil microorganisms. Indicator species analyses showed a vertical variation with a higher importance of saprotrophic taxa in the upper soil layer (0–10 cm) compared to the soil at a depth of 10–20 cm, supporting our second hypothesis. In line with our third hypothesis, bacterial community composition was strongly affected by tree distance, which might be due to higher fine root biomass near spruce trunks. Furthermore, bacterial community composition showed stronger seasonal variation under deciduous trees versus evergreen trees. This pattern was not found when analyzing fungal community composition, which is in contrast to our forth hypothesis. Noteworthy, soil fungal communities under spruce seem to be susceptible to seasonal changes. Overall, our results indicate that trees influence the spatial variation of bacteria and fungi, but their diverse patterns in stem flow, measured by pH change, seem to have a minor impact. Furthermore, the study indicates that soil pH and tree species (European beech or Norway spruce) have a stronger impact on soil bacterial and fungal community composition than soil depth, season or distance from tree trunk.
Additional studies considering root architecture and exudation patterns as well as the influence of tree canopy on the spatial distribution of leaf litter fall are necessary to further elucidate interactions between trees and soil microbes. Besides studies allowing analysis of the proportional importance of factors such as tree species, tree distance, or season, and their mechanisms for interaction, experimental designs focusing on effects of single factors are required to gain more comprehensive understanding on microbial community variation in forest soil. Furthermore, more direct proof is needed to ascertain functional roles of microbes such as Acidobacteria in soil surrounding beech and spruce. For instance, stable isotope probing could be used to identify bacteria or fungi involved in litter degradation.
Author Contributions
MS, FB, RD, and TW designed the study; HN, KG, IS, BP, KK, and GC carried out field and laboratory work; HN, KG, IS, and KK prepared and analyzed the data; all authors interpreted the results and wrote the paper.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank the managers of the three Exploratories, Kirsten Reichel-Jung, Swen Renner, Katrin Hartwich, Sonja Gockel, Kerstin Wiesner, and Martin Gorke for their work in maintaining the plot and project infrastructure; Christiane Fischer and Simone Pfeiffer for giving support through the central office, Michael Owonibi for managing the central data base, and Markus Fischer, Eduard Linsenmair, Dominik Hessenmöller, Jens Nieschulze, Daniel Prati, Ernst-Detlef Schulze, Wolfgang W. Weisser, and the late Elisabeth Kalko for their role in setting up the Biodiversity Exploratories project. The work was funded by the DFG Priority Program 1374 “Infrastructure-Biodiversity-Exploratories” (DA 374/6-1, Core Project 10—SCHR 1181/2-2 and Core Project 11—BU 941/22-1). Field work permits were issued by the responsible state environmental office of Thüringen (according to § 72 BbgNatSchG). This work was further supported by Helmholtz Impulse and Networking Fund through Helmholtz Interdisciplinary Graduate School for Environmental Research (HIGRADE). We kindly thank Beatrix Schnabel for her help while sampling, lab work and together with Melanie Günther and Sigrid Härtling for 454 sequencing. Furthermore, we thank Sandra Klemmer, Theresa Klötzing, Jessica Schäfer, Frederic Van Broeck, Maximilian Frei, and Steffen Both for their field and/or lab assistance and Carla Porges for the drawings of our sampling design. Additionally, we acknowledge support by the DFG and the Open Access Publication Funds of the Göttingen University.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmicb.2016.02067/full#supplementary-material
Table S1. Information on selected trees including tree position, tree age, tree trunk circumference, and sampling direction away from the tree trunk in May and November 2012.
Table S2. Multivariate analysis of variance based on weighted UniFrac distances of whole bacterial and fungal community composition.
Table S3. Multivariate analysis of variance based on weighted UniFrac distances of bacterial and fungal community composition under beech or spruce.
Table S4. Analysis of covariance to test the impact of tree replicate, depth, season and distance from the tree trunk on soil chemical parameters under trees.
Table S5. Results of indicator species analysis showing potential indicative OTUs for tree species, sampling depths, sampling distances and sampling season under trees.
Table S6. Air temperature, soil temperature, and soil water content data.
Figure S1. Rarefaction curves indicating the observed numbers of OTUs at a genetic distance of 3%. Samples derived from soil surrounding beech and samples collected under spruce are represented by brown and green color, respectively.
Figure S2. Line plots showing soil bacterial and fungal diversity as assessed by Shannon index at 3% genetic distance under beech in (A) early summer and (C) autumn, and under spruce in (B) early summer and (D) autumn.
References
Augusto, L., Ranger, J., Binkley, D., and Rothe, A. (2002). Impact of several common tree species of European temperate forests on soil fertility. Ann. For. Sci. 59, 233–253. doi: 10.1051/forest:2002020
Ayres, E., Steltzer, H., Berg, S., Wallenstein, M. D., Simmons, B. L., and Wall, D. H. (2009). Tree species traits influence soil physical, chemical, and biological properties in high elevation forests. PLoS ONE 4:e5964. doi: 10.1371/journal.pone.0005964
Baldrian, P., Kolařík, M., Stursová, M., Kopecký, J., Valášková, V., Větrovský, T., et al. (2012). Active and total microbial communities in forest soil are largely different and highly stratified during decomposition. ISME J. 6, 248–258. doi: 10.1038/ismej.2011.95
Benson, D. A., Clark, K., Karsch-Mizrachi, I., Lipman, D. J., Ostell, J., and Sayers, E. W. (2015). GenBank. Nucleic Acids Res. 43, D30–D35. doi: 10.1093/nar/gku1216
Berger, T. W., and Berger, P. (2012). Greater accumulation of litter in spruce (Picea abies) compared to beech (Fagus sylvatica) stands is not a consequence of the inherent recalcitrance of needles. Plant Soil 358, 349–369. doi: 10.1007/s11104-012-1165-z
Boch, S., Prati, D., Müller, J., Socher, S., Baumbach, H., Buscot, F., et al. (2013). High plant species richness indicates management-related disturbances rather than the conservation status of forests. Basic Appl. Ecol. 14, 496–505. doi: 10.1016/j.baae.2013.06.001
Bragg, L., Stone, G., Imelfort, M., Hugenholtz, P., and Tyson, G. W. (2012). Fast, accurate error-correction of amplicon pyrosequences using Acacia. Nat. Methods 9, 425–426. doi: 10.1038/nmeth.1990
Branco, S., Bruns, T. D., and Singleton, I. (2013). Fungi at a small scale: spatial zonation of fungal assemblages around single trees. PLoS ONE 8:e78295. doi: 10.1371/journal.pone.0078295
Broszat, M., Nacke, H., Blasi, R., Siebe, C., Huebner, J., Daniel, R., et al. (2014). Wastewater irrigation increases the abundance of potentially harmful gammaproteobacteria in soils in Mezquital Valley, Mexico. Appl. Environ. Microbiol. 80, 5282–5291. doi: 10.1128/AEM.01295-14
Caporaso, J. G., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman, F. D., Costello, E. K., et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335–336. doi: 10.1038/nmeth.f.303
Cesarz, S., Fender, A. C., Beyer, F., Valtanen, K., Pfeiffer, B., Gansert, D., et al. (2013). Roots from beech (Fagus sylvatica L.) and ash (Fraxinus excelsior L.) differentially affect soil microorganisms and carbon dynamics. Soil Biol. Biochem. 61, 23–32. doi: 10.1016/j.soilbio.2013.02.003
Coleman, D. C., and Crossley, D. A. (1996). Fundamentals of Soil Ecology. New York, NY: Academic Press.
Cong, J., Yang, Y., Liu, X., Lu, H., Liu, X., Zhou, J., et al. (2015). Analyses of soil microbial community compositions and functional genes reveal potential consequences of natural forest succession. Sci. Rep. 5:10007. doi: 10.1038/srep10007
Crowther, T. W., Glick, H. B., Covey, K. R., Bettigole, C., Maynard, D. S., Thomas, S. M., et al. (2015). Mapping tree density at a global scale. Nature 525, 201–205. doi: 10.1038/nature14967
De Cáceres, M., and Legendre, P. (2009). Associations between species and groups of sites: indices and statistical inference. Ecology 90, 3566–3574. doi: 10.1890/08-1823.1
Edgar, R. C. (2010). Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461. doi: 10.1093/bioinformatics/btq461
Edgar, R. C., Haas, B. J., Clemente, J. C., Quince, C., and Knight, R. (2011). UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27, 2194–2200. doi: 10.1093/bioinformatics/btr381
Eilers, K. G., Debenport, S., Anderson, S., and Fierer, N. (2012). Digging deeper to find unique microbial communities: the strong effect of depth on the structure of bacterial and archaeal communities in soil. Soil Biol. Biochem. 50, 58–65. doi: 10.1016/j.soilbio.2012.03.011
Erlacher, A., Cernava, T., Cardinale, M., Soh, J., Sensen, C. W., Grube, M., et al. (2015). Rhizobiales as functional and endosymbiontic members in the lichen symbiosis of Lobaria pulmonaria L. Front. Microbiol. 6:53. doi: 10.3389/fmicb.2015.00053
Ettema, C. H., and Wardle, D. A. (2002). Spatial soil ecology. Trends Ecol. Evol. 17, 177–183. doi: 10.1016/S0169-5347(02)02496-5
Fender, A. C., Gansert, D., Jungkunst, H. F., Fiedler, S., Beyer, F., Schützenmeister, K., et al. (2013). Root-induced tree species effects on the source/sink strength for greenhouse gases (CH4, N2O and CO2) of a temperate deciduous forest soil. Soil Biol. Biochem. 57, 587–597. doi: 10.1016/j.soilbio.2012.08.004
Fischer, M., Bossdorf, O., Gockel, S., Hänsel, F., Hemp, A., Hessenmöller, D., et al. (2010). Implementing large-scale and long-term functional biodiversity research: the biodiversity exploratories. Basic Appl. Ecol. 11, 473–485. doi: 10.1016/j.baae.2010.07.009
García-Fraile, P., Benada, O., Cajthaml, T., Baldrian, P., and Lladó, S. (2015). Terracidiphilus gabretensis gen. nov., sp. nov., an abundant and active forest soil Acidobacterium important in organic matter transformation. Appl. Environ. Microbiol. 82, 560–569. doi: 10.1128/AEM.03353-15
Gardes, M., and Bruns, T. D. (1993). ITS primers with enhanced specificity for basidiomycetes – application to the identification of mycorrhizae and rusts. Mol. Ecol. 2, 113–118. doi: 10.1111/j.1365-294X.1993.tb00005.x
Geßler, A., Schneider, S., Weber, P., Hanemann, U., and Rennenberg, H. (1998). Soluble N compounds in trees exposed to high loads of N: a comparison between the roots of Norway spruce (Picea abies) and beech (Fagus sylvatica) trees grown under field conditions. New Phytol. 138, 385–399. doi: 10.1046/j.1469-8137.1998.00134.x
Goldfarb, K. C., Karaoz, U., Hanson, C. A., Santee, C. A., Bradford, M. A., Treseder, K. K., et al. (2011). Differential growth responses of soil bacterial taxa to carbon substrates of varying chemical recalcitrance. Front. Microbiol. 2:94. doi: 10.3389/fmicb.2011.00094
Goldmann, K., Schöning, I., Bucot, F., and Wubet, T. (2015). Forest management type influences diversity and community composition of soil fungi across temperate forest ecosystems. Front. Microbiol. 6:1300. doi: 10.3389/fmicb.2015.01300
Grebenc, T., and Kraigher, H. (2007). Types of ectomycorrhiza of mature beech and spruce at ozone-fumigated and control forest plots. Environ. Monit. Assess. 128, 47–59. doi: 10.1007/s10661-006-9414-3
Hammer, Ø., Harper, D. A. T., and Ryan, P. D. (2001). PAST: paleontological statistics software package for education and data analysis. Palaeontologia Electronica 4, 1–9. Available online at: http://palaeo-electronica.org/2001_1/past/issue1_01.htm
Hanewinkel, M., Cullmann, D. A., Schelhaas, M. J., Nabuurs, G. J., and Zimmermann, N. E. (2013). Climate change may cause severe loss in the economic value of European forest land. Nat. Clim. Change 3, 203–207. doi: 10.1038/nclimate1687
Hansel, C. M., Fendorf, S., Jardine, P. M., and Francis, C. A. (2008). Changes in bacterial and archaeal community structure and functional diversity along a geochemically variable soil profile. Appl. Environ. Microbiol. 74, 1620–1633. doi: 10.1128/AEM.01787-07
Houghton, K. M., Morgan, X. C., Lagutin, K., MacKenzie, A. D., Vyssotskii, M., Mitchell, K. A., et al. (2015). Thermorudis pharmacophila sp. nov., a novel member of the class Thermomicrobia isolated from geothermal soil, and emended descriptions of Thermomicrobium roseum, Thermomicrobium carboxidum, Thermorudis peleae and Sphaerobacter thermophilus. Int. J. Syst. Evol. Microbiol. 65, 4479–4487. doi: 10.1099/ijsem.000598
Huang, J., Sheng, X., He, L., Huang, Z., Wang, Q., and Zhang, Z. (2013). Characterization of depth-related changes in bacterial community compositions and functions of a paddy soil profile. FEMS Microbiol. Lett. 347, 33–42. doi: 10.1111/1574-6968.12218
Hug, L. A., Castelle, C. J., Wrighton, K. C., Thomas, B. C., Sharon, I., Frischkorn, K. R., et al. (2013). Community genomic analyses constrain the distribution of metabolic traits across the Chloroflexi phylum and indicate roles in sediment carbon cycling. Microbiome 1:22. doi: 10.1186/2049-2618-1-22
Ishida, T. A., Nara, K., and Hogetsu, T. (2007). Host effects on ectomycorrhizal fungal communities: insight from eight host species in mixed conifer-broadleaf forests. New Phytol. 174, 430–440. doi: 10.1111/j.1469-8137.2007.02016.x
Jiang, Y., Chen, C., Xu, Z., and Liu, Y. (2012). Effects of single and mixed species forest ecosystems on diversity and function of soil microbial community in subtropical China. J. Soils Sediments 12, 228–240. doi: 10.1007/s11368-011-0442-4
Johansson, M., and Marklund, E. (1980). Antagonists of Fomes annosus in the rhizosphere of grey alder (Alnus incana) and Norway spruce (Picea abies). Eur. J. For. Pathol. 10, 385–395. doi: 10.1111/j.1439-0329.1980.tb00056.x
Johnson, M. S., and Lehmann, J. (2006). Double-funneling of trees: stemflow and root-induced preferential flow. Ecoscience 13, 324–333. doi: 10.2980/i1195-6860-13-3-324.1
Johnson, M., Zaretskaya, I., Raytselis, Y., Merezhuk, Y., McGinnis, S., and Madden, T. L. (2008). NCBI BLAST: a better web interface. Nucleic Acids Res. 36, W5–W9. doi: 10.1093/nar/gkn201
Kadowaki, K., Sato, H., Yamamoto, S., Tanabe, A. S., Hidaka, A., and Toju, H. (2014). Detection of the horizontal spatial structure of soil fungal communities in a natural forest. Popul. Ecol. 56, 301–310. doi: 10.1007/s10144-013-0424-z
Kaiser, C., Koranda, M., Kitzler, B., Fuchslueger, L., Schnecker, J., Schweiger, P., et al. (2010). Belowground carbon allocation by trees drives seasonal patterns of extracellular enzyme activities by altering microbial community composition in a beech forest soil. New Phytol. 187, 843–858. doi: 10.1111/j.1469-8137.2010.03321.x
Kämpfer, P., Young, C. C., Arun, A. B., Shen, F. T., Jäckel, U., Rosselló-Mora, R., et al. (2006). Pseudolabrys taiwanensis gen. nov., sp. nov., an alphaproteobacterium isolated from soil. Int. Syst. Evol. Microbiol. 56, 2469–2472. doi: 10.1099/ijs.0.64124-0
Koch, A. S., and Matzner, E. (1993). Heterogeneity of soil and soil solution chemistry under Norway Spruce (Picea abies Karst.) and European Beech (Fagus silvatica L.) as influenced by distance from the stem basis. Plant Soil 151, 227–237. doi: 10.1007/BF00016288
Kõljalg, U., Nilsson, R. H., Abarenkov, K., Tedersoo, L., Taylor, A. F., Bahram, M., et al. (2013). Towards a unified paradigm for sequence-based identification of fungi. Mol. Ecol. 22, 5271–5277. doi: 10.1111/mec.12481
Korkama-Rajala, T., Müller, M. M., and Pennanen, T. (2008). Decomposition and fungi of needle litter from slow- and fast-growing Norway spruce (Picea abies) clones. Microb. Ecol. 56, 76–89. doi: 10.1007/s00248-007-9326-y
Lauber, C. L., Hamady, M., Knight, R., and Fierer, N. (2009). Pyrosequencing-based assessment of soil pH as a predictor of soil bacterial community structure at the continental scale. Appl. Environ. Microbiol. 75, 5111–5120. doi: 10.1128/AEM.00335-09
Lauber, C. L., Strickland, M. S., Bradford, M. A., and Fierer, N. (2008). The influence of soil properties on the structure of bacterial and fungal communities across land-use types. Soil Biol. Biochem. 40, 2407–2415. doi: 10.1016/j.soilbio.2008.05.021
Lejon, D. P., Chaussod, R., Ranger, J., and Ranjard, L. (2005). Microbial community structure and density under different tree species in an acid forest soil (Morvan, France). Microb. Ecol. 50, 614–625. doi: 10.1007/s00248-005-5130-8
Li, W., and Godzik, A. (2006). Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 22, 1658–1659. doi: 10.1093/bioinformatics/btl158
Lin, W. R., Wang, P. H., Chen, W. C., Lai, C. M., and Winder, R. S. (2016). Responses of soil fungal populations and communities to the thinning of Cryptomeria japonica Forests. Microbes Environ. 31, 19–26. doi: 10.1264/jsme2.ME15127
Liu, Z., Lozupone, C., Hamady, M., Bushman, F. D., and Knight, R. (2007). Short pyrosequencing reads suffice for accurate microbial community analysis. Nucleic Acids Res. 35:e120. doi: 10.1093/nar/gkm541
Štursová, M., Žifčáková, L., Leigh, M. B., Burgess, R., and Baldrian, P. (2012). Cellulose utilization in forest litter and soil: identification of bacterial and fungal decomposers. FEMS Microbiol. Ecol. 80, 735–746. doi: 10.1111/j.1574-6941.2012.01343.x
López-Mondéjar, R., Voříšková, J., Vĕtrovský, T., and Baldrian, P. (2015). The bacterial community inhabiting temperate deciduous forests is vertically stratified and undergoes seasonal dynamics. Soil Biol. Biochem. 87, 43–50. doi: 10.1016/j.soilbio.2015.04.008
Lozupone, C., Lladser, M. E., Knights, D., Stombaugh, J., and Knight, R. (2011). UniFrac: an effective distance metric for microbial community comparison. ISME J. 5, 169–172. doi: 10.1038/ismej.2010.133
Martin, M. (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 17, 10–12. doi: 10.14806/ej.17.1.200
McGuire, K. L., Allison, S. D., Fierer, N., and Treseder, K. K. (2013). Ectomycorrhizal-dominated boreal and tropical forests have distinct fungal communities, but analogous spatial patterns across soil horizons. PLoS ONE 8:e68278. doi: 10.1371/journal.pone.0068278
McGuire, K. L., Bent, E., Borneman, J., Majumder, A., Allison, S. D., and Tresederi, K. K. (2010). Functional diversity in resource use by fungi. Ecology 91, 2324–2332. doi: 10.1890/09-0654.1
Miyamoto, Y., Sakai, A., Hattori, M., and Nara, K. (2015). Strong effect of climate on ectomycorrhizal fungal composition: evidence from range overlap between two mountains. ISME J. 9, 1870–1879. doi: 10.1038/ismej.2015.8
Moll, J., Goldmann, K., Kramer, S., Hempel, S., Kandeler, E., Marhan, S., et al. (2015). Resource type and availability regulate fungal communities along arable soil profiles. Microb. Ecol. 70, 390–399. doi: 10.1007/s00248-015-0569-8
Morales, S. E., Cosart, T. F., Johnson, J. V., and Holben, W. E. (2009). Extensive phylogenetic analysis of a soil bacterial community illustrates extreme taxon evenness and the effects of amplicon length, degree of coverage, and DNA fractionation on classification and ecological parameters. Appl. Environ. Microbiol. 75, 668–675. doi: 10.1128/AEM.01757-08
Nacke, H., Thürmer, A., Wollherr, A., Will, C., Hodac, L., Herold, N., et al. (2011). Pyrosequencing-based assessment of bacterial community structure along different management types in German forest and grassland soils. PLoS ONE 6:e17000. doi: 10.1371/journal.pone.0017000
Nagy, L. G., Petkovits, T., Kovacs, G. M., Voigt, K., Vagvolgyi, C., and Papp, T. (2011). Where is the unseen fungal diversity hidden? A study of Mortierella reveals a large contribution of reference collections to the identification of fungal environmental sequences. New Phytol. 191, 789–794. doi: 10.1111/j.1469-8137.2011.03707.x
Nakatsu, C. H., Carmosini, N., Baldwin, B., Beasley, F., Kourtev, P., and Konopka, A. (2005). Soil microbial community responses to additions of organic carbon substrates and heavy metals (Pb and Cr). Appl. Environ. Microbiol. 71, 7679–7689. doi: 10.1128/AEM.71.12.7679-7689.2005
Nykvist, N. (1963). Leaching and decomposition of water-soluble organic substances from different types of leaf and needle litter. Stud. For. Suec. 3, 1–31.
Oksanen, J., Blanchet, F., Kindt, R., Legendre, P., Minchin, P., O'Hara, R., et al (2016). vegan: Community Ecology Package. Oulu: University of Oulu. Available online at: http://CRAN.R-project.org/package=vegan
Pankratov, T. A., Tindall, B. J., Liesack, W., and Dedysh, S. N. (2007). Mucilaginibacter paludis gen. nov., sp. nov. and Mucilaginibacter gracilis sp. nov., pectin-, xylan- and laminarin-degrading members of the family Sphingobacteriaceae from acidic Sphagnum peat bog. Int. J. Syst. Evol. Microbiol. 57, 2349–2354. doi: 10.1099/ijs.0.65100-0
Petritan, I. C., von Lupke, B., and Petritan, A. M. (2011). Fine roots of overstory Norway spruce (Picea abies): distribution and influence on growth of underplanted beech (Fagus sylvatica) and Douglas-fir (Pseudotsuga menziesii) saplings. For. Syst. 20, 407–419. doi: 10.5424/fs/20112003/11136
Pfeiffer, B., Fender, A. C., Lasota, S., Hertel, D., Jungkunst, H. F., and Daniel, R. (2013). Leaf litter is the main driver for changes in bacterial community structures in the rhizosphere of ash and beech. Appl. Soil Ecol. 72, 150–160. doi: 10.1016/j.apsoil.2013.06.008
Polishook, J. D., Bills, G. F., and Lodge, D. J. (1996). Microfungi from decaying leaves of two rain forest trees in Puerto Rico. J. Ind. Microbiol. Biotechnol. 17, 284–294. doi: 10.1007/BF01574703
Prescott, C. E. (2010). Litter decomposition: what controls it and how can we alter it to sequester more carbon in forest soils? Biogeochemistry 101, 133–149. doi: 10.1007/s10533-010-9439-0
Priha, O., and Smolander, A. (1997). Microbial biomass and activity in soil and litter under Pinus sylvestris, Picea abies and Betula pendula at originally similar field afforestation sites. Biol. Fert. Soils 24, 45–51. doi: 10.1007/BF01420219
Pruesse, E., Quast, C., Knittel, K., Fuchs, B. M., Ludwig, W., Peplies, J., et al. (2007). SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res. 35, 7188–7196. doi: 10.1093/nar/gkm864
Purahong, W., Hoppe, B., Kahl, T., Schloter, M., Schulze, E. D., Bauhus, J., et al. (2014). Changes within a single land-use category alter microbial diversity and community structure: molecular evidence from wood-inhabiting fungi in forest ecosystems. J. Environ. Manage. 139, 109–119. doi: 10.1016/j.jenvman.2014.02.031
Raz-Yaseef, N., Rotenberg, E., and Yakir, D. (2010). Effects of spatial variations in soil evaporation caused by tree shading on water flux partitioning in a semi-arid pine forest. Agric. For. Meteorol. 150, 454–462. doi: 10.1016/j.agrformet.2010.01.010
R Development Core Team (2015). R: A Language and Environment for Statistical Computing. Vienna: Foundation for Statistical Computing.
Rosselló-Mora, R., and Amann, R. (2001). The species concept for prokaryotes. FEMS Microbiol. Rev. 25, 39–67. doi: 10.1111/j.1574-6976.2001.tb00571.x
Saetre, P., and Bååth, E. (2000). Spatial variation and patterns of soil microbial community structure in a mixed spruce-birch stand. Soil Biol. Biochem. 32, 909–917. doi: 10.1016/S0038-0717(99)00215-1
Scattolin, L., Montecchio, L., Mosca, E., and Agerer, R. (2008). Vertical distribution of the ectomycorrhizal community in the top soil of Norway spruce stands. Eur. J. For. Res. 127, 347–357. doi: 10.1007/s10342-008-0209-7
Schloss, P. D., Westcott, S. L., Ryabin, T., Hall, J. R., Hartmann, M., Hollister, E. B., et al. (2009). Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 75, 7537–7541. doi: 10.1128/AEM.01541-09
Schmidt, O. (2013). Holz-und Baumpilze: Biologie, Schäden, Schutz, Nutzen. Heidelberg: Springer-Verlag.
Schweitzer, J. A., Bailey, J. K., Fischer, D. G., LeRoy, C. J., Lonsdorf, E. V., Whitham, T. G., et al. (2008). Plant-soil-microorganism interactions: heritable relationship between plant genotype and associated soil microorganisms. Ecology 89, 773–781. doi: 10.1890/07-0337.1
Shay, P. E., Winder, R. S., and Trofymow, J. A. (2015). Nutrient-cycling microbes in coastal Douglas-fir forests: regional-scale correlation between communities, in situ climate, and other factors. Front. Microbiol. 6:1097. doi: 10.3389/fmicb.2015.01097
Shi, L., Guttenberger, M., Kottke, I., and Hampp, R. (2002). The effect of drought on mycorrhizas of beech (Fagus sylvatica L.): changes in community structure, and the content of carbohydrates and nitrogen storage bodies of the fungi. Mycorrhiza 12, 303–311. doi: 10.1007/s00572-002-0197-2
Stevenson, B. A., Hunter, D. W. F., and Rhodes, P. L. (2014). Temporal and seasonal change in microbial community structure of an undisturbed, disturbed, and carbon-amended pasture soil. Soil Biol. Biochem. 75, 175–185. doi: 10.1016/j.soilbio.2014.04.010
Summerbell, R. C. (2005). Root endophyte and mycorrhizosphere fungi of black spruce, Picea mariana, in a boreal forest habitat: influence of site factors on fungal distributions. Stud. Mycol. 53, 121–145. doi: 10.3114/sim.53.1.121
Tedersoo, L., Bahram, M., Cajthaml, T., Põlme, S., Hiiesalu, I., Anslan, S., et al. (2016). Tree diversity and species identity effects on soil fungi, protists and animals are context dependent. ISME J. 10, 346–362. doi: 10.1038/ismej.2015.116
Thoms, C., Gattinger, A., Jacob, M., Thomas, F. M., and Gleixner, G. (2010). Direct and indirect effects of tree diversity drive soil microbial diversity in temperate deciduous forest. Soil Biol. Biochem. 42, 1558–1565. doi: 10.1016/j.soilbio.2010.05.030
Thoms, C., and Gleixner, G. (2013). Seasonal differences in tree species' influence on soil microbial communities. Soil Biol. Biochem. 66, 239–248. doi: 10.1016/j.soilbio.2013.05.018
Trevors, J. T. (2010). One gram of soil: a microbial biochemical gene library. Antonie van Leeuwenhoek 97, 99–106. doi: 10.1007/s10482-009-9397-5
Urbanová, M., Šnajdr, J., and Baldrian, P. (2015). Composition of fungal and bacterial communities in forest litter and soil is largely determined by dominant trees. Soil Biol. Biochem. 84, 53–64. doi: 10.1016/j.soilbio.2015.02.011
Uroz, S., Oger, P., Tisserand, E., Cébron, A., Turpault, M. P., Buée, M., et al. (2016). Specific impacts of beech and Norway spruce on the structure and diversity of the rhizosphere and soil microbial communities. Sci. Rep. 6:27756. doi: 10.1038/srep27756
van der Heijden, M. G., Bardgett, R. D., and van straalen, N. M. (2008). The unseen majority: soil microbes as drivers of plant diversity and productivity in terrestrial ecosystems. Ecol. Lett. 11, 296–310. doi: 10.1111/j.1461-0248.2007.01139.x
Vázquez-Baeza, Y., Pirrung, M., Gonzalez, A., and Knight, R. (2013). Emperor: a tool for visualizing high-throughput microbial communitiy data. Gigascience 2:16. doi: 10.1186/2047-217X-2-16
Velmala, S. M., Rajala, T., Haapanen, M., Taylor, A. F., and Pennanen, T. (2013). Genetic host-tree effects on the ectomycorrhizal community and root characteristics of Norway spruce. Mycorrhiza 23, 21–33. doi: 10.1007/s00572-012-0446-y
Voříšková, J., Brabcov,á, V., Cajthaml, T., and Baldrian, P. (2014). Seasonal dynamics of fungal communities in a temperate oak forest soil. New Phytol. 201, 269–278. doi: 10.1111/nph.12481.
Wäldchen, J., Schöning, I., Mund, M., Schrumpf, M., Bock, S., Herold, N., et al. (2012). Estimation of clay content from easily measurable water content of air-dried soil. J. Plant Nutr. Soil Sci. 175, 367–376. doi: 10.1002/jpln.201100066
Wäldchen, J., Schulze, E. D., Mund, M., and Winkler, B. (2011). Der Einfluss politischer, rechtlicher und wirtschaftlicher Rahmenbedingungen des 19. Jahrhunderts auf die Bewirtschaftung der Wälder im Hainich-Dün-Gebiet (Nordthüringen). Forstarchiv 82, 35–47. doi: 10.2376/0300-4112-82-35
Wang, Y., and Qian, P. Y. (2009). Conservative fragments in bacterial 16S rRNA genes and primer design for 16S ribosomal DNA amplicons in metagenomic studies. PLoS ONE 4:e7401. doi: 10.1371/journal.pone.0007401
Ward, N. L., Challacombe, J. F., Janssen, P. H., Henrissat, B., Coutinho, P. M., Wu, M., et al. (2009). Three genomes from the phylum Acidobacteria provide insight into the lifestyles of these microorganisms in soils. Appl. Environ. Microbiol. 75, 2046–2056. doi: 10.1128/AEM.02294-08
Weber, P., Stoermer, H., Geßler, A., Schneider, S., von Sengbusch, D., Hanemann, U., et al. (1998). Metabolic responses of Norway spruce (Picea abies) trees to long-term forest management practices and acute (NH4)2SO4 fertilization: transport of soluble non-protein nitrogen compounds in xylem and phloem. New Phytol. 140, 461–475. doi: 10.1111/j.1469-8137.1998.00285.x
White, T., Brans, T., Lee, S., and Taylor, J. (1990). “Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics,” in PCR Protocols: A Guide to Methods and Applications, eds M. A. Innis, D. H. Gelfand and J. J. Sninsky (San Diego, CA: Academic Press), 315.
Will, C., Thürmer, A., Wollherr, A., Nacke, H., Herold, N., Schrumpf, M., et al. (2010). Horizon-specific bacterial community composition of German grassland soils, as revealed by pyrosequencing-based analysis of 16S rRNA genes. Appl. Environ. Microbiol. 76, 6751–6759. doi: 10.1128/AEM.01063-10.
Wubet, T., Christ, S., Schöning, I., Boch, S., Gawlich, M., Schnabel, B., et al. (2012). Differences in soil fungal communities between European beech (Fagus sylvatica L.) dominated forests are related to soil and understory vegetation. PLoS ONE 7:e47500. doi: 10.1371/journal.pone.0047500
Wuczkowski, M., Druzhinina, I., Gherbawy, Y., Klug, B., Prillinger, H., and Kubicek, C. P. (2003). Species pattern and genetic diversity of Trichoderma in a mid-European, primeval floodplain-forest. Microbiol. Res. 158, 125–133. doi: 10.1078/0944-5013-00193
Yarwood, S. A., Bottomley, P. J., and Myrold, D. D. (2010). Soil microbial communities associated with Douglas-fir and red alder stands at high- and low-productivity forest sites in Oregon, USA. Microb. Ecol. 60, 606–617. doi: 10.1007/s00248-010-9675-9
Zechmeister-Boltenstern, S., Michel, K., and Pfeffer, M. (2011). Soil microbial community structure in European forests in relation to forest type and atmospheric nitrogen deposition. Plant Soil 343, 37–50. doi: 10.1007/s11104-010-0528-6
Keywords: tree species, soil depth, horizontal distance from tree trunk, seasons, soil properties, soil microbial community structure, bacterial 16S rRNA gene, fungal ITS DNA
Citation: Nacke H, Goldmann K, Schöning I, Pfeiffer B, Kaiser K, Castillo-Villamizar GA, Schrumpf M, Buscot F, Daniel R and Wubet T (2016) Fine Spatial Scale Variation of Soil Microbial Communities under European Beech and Norway Spruce. Front. Microbiol. 7:2067. doi: 10.3389/fmicb.2016.02067
Received: 15 August 2016; Accepted: 07 December 2016;
Published: 22 December 2016.
Edited by:
Tim Daniell, James Hutton Institute, UKReviewed by:
Christopher Blackwood, Kent State University, USARichard S. Winder, Natural Resources Canada, Canada
Christina Hazard, University of Lyon, France
Copyright © 2016 Nacke, Goldmann, Schöning, Pfeiffer, Kaiser, Castillo-Villamizar, Schrumpf, Buscot, Daniel and Wubet. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Heiko Nacke, hnacke@gwdg.de
†These authors have contributed equally to this work.