 Yuan Gao1
Yuan Gao1 Xin Yang
Xin Yang Chun-Ren Wang
Chun-Ren Wang- 1College of Animal Science and Veterinary Medicine, Heilongjiang Bayi Agricultural University, Daqing, China
- 2State Key Laboratory of Agricultural Microbiology, College of Veterinary Medicine, Huazhong Agricultural University, Wuhan, China
- 3College of Life Science and Biotechnology, Heilongjiang Bayi Agricultural University, Daqing, China
Equine strongyles, the significant nematode pathogens of horses, are characterized by high quantities and species abundance, but classification of this group of parasitic nematodes is debated. Mitochondrial (mt) genome DNA data are often used to address classification controversies. Thus, the objectives of this study were to determine the complete mt genomes of three Cyathostominae nematode species (Cyathostomum catinatum, Cylicostephanus minutus, and Poteriostomum imparidentatum) of horses and reconstruct the phylogenetic relationship of Strongylidae with other nematodes in Strongyloidea to test the hypothesis that Triodontophorus spp. belong to Cyathostominae using the mt genomes. The mt genomes of Cy. catinatum, Cs. minutus, and P. imparidentatum were 13,838, 13,826, and 13,817 bp in length, respectively. Complete mt nucleotide sequence comparison of all Strongylidae nematodes revealed that sequence identity ranged from 77.8 to 91.6%. The mt genome sequences of Triodontophorus species had relatively high identity with Cyathostominae nematodes, rather than Strongylus species of the same subfamily (Strongylinae). Comparative analyses of mt genome organization for Strongyloidea nematodes sequenced to date revealed that members of this superfamily possess identical gene arrangements. Phylogenetic analyses using mtDNA data indicated that the Triodontophorus species clustered with Cyathostominae species instead of Strongylus species. The present study first determined the complete mt genome sequences of Cy. catinatum, Cs. minutus, and P. imparidentatum, which will provide novel genetic markers for further studies of Strongylidae taxonomy, population genetics, and systematics. Importantly, sequence comparison and phylogenetic analyses based on mtDNA sequences supported the hypothesis that Triodontophorus belongs to Cyathostominae.
Introduction
Equine strongyles, a large group of intestinal nematodes that belong to members of Equidae, are classified into two subfamilies, Strongylinae (large strongyles) and Cyathostominae (small strongyles), based on worm size and capsule mouth shape (Lichtenfels et al., 2008; Traversa et al., 2010). Although equine strongyles are the significant nematode pathogens of horses, information on these nematodes is limited to their morphology, prevalence, and disease control and prevention (Bu et al., 2009; Lyons et al., 2011; Mughini et al., 2011; Morariu et al., 2016; Singh et al., 2016). Intestinal nematodes of Cyathostominae, a ubiquitous parasitic nematode species, inhabit the large intestine of infested equines with a high prevalence, especially because of the reduced prevalence of Strongylus spp. and spread of cyathostomin anthelmintic-resistant populations (Reinemeyer, 1986; Traversa et al., 2010). Small strongyles can infect virtually any horse, with symptoms of anorexia, weight loss, poor hair coat, lethargy with disordered intestinal motility, and some with inflammatory enteropathy caused by adult Cyathostominae, especially as a result of emergence of enormous numbers of larvae from the lining of the large intestine with a high mortality rate of up to 50% caused by larval stages (Reinemeyer, 1986; Love et al., 1999; Corning, 2009; Traversa et al., 2010).
The traditional classification of equine strongyles was primarily based on morphological characteristics, but some researchers believed that the classification of Strongylidae (including the separation of Strongylinae and Cyathostominae) based on differences in the size and shape of the buccal capsule was arbitrary (Durette-Desset et al., 1994; Lichtenfels et al., 1998). However, reconstruction of the systematic relationships using the first and second internal transcribed spacers (ITS1 and ITS2, respectively) of 30 equine strongyles species revealed that Triodontophorus serratus, which was previously classified into Strongylinae based on morphology, clustered with Cyathostominae (Hung et al., 2000).
Mitochondrial (mt) genome sequences provide effective and reliable molecular markers for various types of evolutionary studies of parasites because of their strict maternal inheritance, apparent lack of recombination, rapid evolutionary rate, and comparatively conserved genomic structure (Gissi et al., 2008; Jia et al., 2010; Tian et al., 2015; Zhang et al., 2015; Guo et al., 2016; Liu et al., 2016), especially for classification of higher taxonomic levels. For example, the mt genome provided evidence that Orientobilharzia turkestanicum belongs to the genus Schistosoma, and is phylogenetically closer to the African schistosome group than to the Asian schistosome group (Wang et al., 2011). In addition, phylogenetic analyses of both nucleotide and amino acid sequence data of mt genome supported the hypothesis that Dicrocoelium chinensis and D. dendriticum are genetically distinct species (Liu et al., 2014).
Therefore, the objectives of this study were to: (1) determine the complete mt genome sequences of the three nematodes Cyathostomum catinatum, Cylicostephanus minutus, and Poteriostomum imparidentatum; (2) analyze and compare the mt genomes of Strongylidae nematodes; and (3) reconstruct the phylogenetic relationship of Strongylidae with other nematodes in Strongyloidea based on the mtDNA data to test the hypothesis that Triodontophorus belongs to Cyathostominae.
Materials and Methods
Ethics Approval
This study was approved by the Animal Ethics Committee of Heilongjiang Bayi Agricultural University. Horses used for the study were handled in accordance with good animal practice, as defined by the Animal Ethics Procedures and Guidelines of the People's Republic of China.
Parasites, DNA Extraction, Genome Amplification, and Sequence Analyses
Adult nematode Cy. catinatum, Cs. minutus, and P. imparidentatum were collected from the large intestine of naturally infected horses from a slaughter house in Daqing, Heilongjiang Province, China. Individual worms were identified to species based on their morphological characteristics and predilection sites (Lichtenfels et al., 2008). Under the microscope, the morphological characteristics of Cy. catinatum including the clear mouth collar, buccal cavity wide in the front and narrow in the back and its width greater than its depth, short dorsal lobe of male bursa and the foot-shaped tail of female could be observed (see Figure S1). Cs. minutus is small with a short mouth collar, clear and long submedian papilla, buccal cavity deeper than wide, dorsal gutter elongate reaching half the depth of buccal capsule, short dorsal lobe of male bursa, and straight tails for females (see Figure S2). P. imparidentatum are relatively large and have a clear internal and external leaf-crown, high mouth collar, fine and sharp submedian papilla, width of buccal capsule greater than depth, and short and wide dorsal lobe of male bursa (see Figure S3). The detailed data are listed in Table 1.
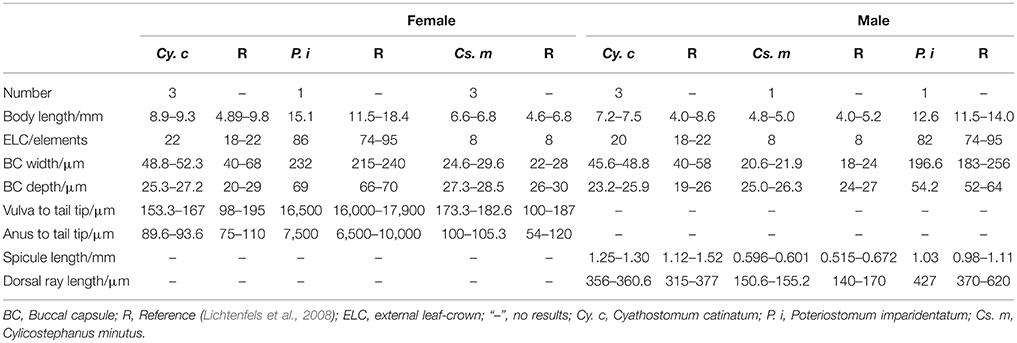
Table 1. The detail information of morphological characteristics of the three nematodes.
Subsequently, total genomic DNA was extracted from individual worms using TIANamp Genomic DNA Kit (TIANGEN Biotech, Beijing, China) according to the manufacturer's instructions. Parasite species were determined by PCR amplification of the ITS sequences of Cy. catinatum, Cs. minutus, P. imparidentatum, using the universal primers NC5 (5′- GTA GGT GAA CCT GCG GAA GGA TCA TT -3′) and NC2 (5′- TTA GTT TCT TTT CCT CCG CT -3′) reported by Gasser et al. (2008). The ITS sequences of Cy. catinatum, Cs. minutus, and P. imparidentatum obtained in the present study had 99.8, 99.7, and 99.2% identities to the corresponding available sequences in GenBank (Cs. minutus, KM085361.1; P. imparidentatum, KP693433.1; Cy. catinatum, KF850626.1, respectively), which confirmed the identity of the examined three species. The specific primers for mt genomes of the three worms were designed based on the relatively conserved mt sequences of the Strongylidae horse parasites available in GenBank (Table S1). PCR cycling conditions used to amplify mtDNA of the three nematodes were based on those of a previous report (Gao et al., 2017). After PCR amplification, the positive amplicons were sent to Life Technology Company (Beijing, China) for sequencing. With the aid of bioinformatics software such as MegAlign 5.01, Clustal X 1.83, MEGA 5.0, and tRNAscan-SE 2.0 (http://lowelab.ucsc.edu/tRNAscan-SE/) (Thompson et al., 1997; Burland, 2000; Tamura et al., 2011) and manual analysis, the boundary of each protein-coding gene, transfer RNA (tRNA) gene, and ribosomal RNA (rRNA) gene was determined.
Comparative Analysis with Other Strongylidae Worms
Comparisons were made based on mtDNA size, gene arrangement, percentage of A+T content, and nucleotide and amino acid sequence similarity inferred from individual protein-coding genes among Cy. catinatum, Cs. minutus, P. imparidentatum, and nine other Strongylidae nematodes for which mt genome sequences were available in GenBank, which included Cylicocyclus insigne (NC_013808.1), Cs. goldi (AP017681.1), Cylicocyclus nassatus (KX819273.1), Strongylus equinus (NC_026868.1), S. vulgaris (AP017698.1), T. brevicauda (NC_026729.1), T. nipponicus (NC_031517.1), T. serratus (NC_031516.1), Macropicola ocydromi (NC_023099.1).
Phylogenetic Analyses
Phylogenetic analysis in this study was based on the concatenated amino acid sequences of 12 protein-coding genes of 20 Strongyloidea nematodes available in GenBank from family Chabertiidae: Chabertia erschowi (KF660603), C. ovina (NC_013831); family Cloacinidae: Oesophagostomum asperum (NC_023932.1), O. columbianum (NC_023933.1), O. dentatum (GQ888716), O. quadrispinulatum (NC_014181), Hypodontus macropi (NC_023099.1); family Strongylidae, including two subfamilies, Cyathostominae (Cs. goldi, Cc. insigne, Cc. nassatus, Cy. catinatum, Cs. minutus, and P. imparidentatum) and Strongylinae (S. equinus, S. vulgaris, T. brevicauda, T. nipponicus, T. serratus, M. ocydromi); and family Syngamidae: Syngamus trachea (GQ888718), and family Ancylostomatidae: Ancylostoma duodenale (NC_003415) as an outgroup. Each gene was translated using the invertebrate mitochondrial genetic code in MEGA 5 (Tamura et al., 2011) and ambiguously aligned regions were excluded using the Gblocks Server (http://molevol.cmima.csic.es/castresana/Gblocks_server.html) with less stringent selection. Phylogenetic trees were all reconstructed using Bayesian inference (BI) methods, which were performed using the mixed model in MrBayes 3.1.1 and 1,000,000 metropolis-coupled Markov chain Monte Carlo generations (Ronquist and Huelsenbeck, 2003); maximum parsimony (MP) methods, which were performed using a Fitch criterion (1,000 bootstrap replicates) within PAUP 4.0 Beta 10 (Swofford, 2002); and maximum likelihood (ML) methods (JTT+I+G+F model) using PhyML 3.0 (Guindon and Gascuel, 2003), and bootstrapping was performed using 100 replicates. Phylograms were drawn using Tree View 1.65 (Page, 1996).
Results
General Features of the Three MT Genomes
In the present study, the complete mt genomes of Cy. catinatum, Cs. minutus, and P. imparidentatum were 13,838 bp, 13,826 bp and 13,817 bp in length, respectively (Table 2). The mt genomes of Cy. catinatum and P. imparidentatum are the first reported for both genera. All three mt genomes contained 12 protein-coding genes (nad1–nad6 and nad4L, cox1–cox3, cytb, atp6), 22 tRNA genes, two rRNA genes, and two non-coding regions (Table 2), which were transcribed in the same direction. Moreover, there were 19, 20, and 16 intergenic sequences in the complete circular mt genomes of Cy. catinatum, Cs. minutus, and P. imparidentatum, respectively. The largest intergenic region occurred in Cy. catinatum and was 51 bp in length. The shortest intergenic regions were 1-bp intergenic sequences in all three mt genomes. One, two, and three 1-bp overlaps were present in the mt genomes of Cy. catinatum, Cs. minutus, and P. imparidentatum, respectively. The total A+T contents in the genome sequences ranged from 74.65 to 76.12%, and the A, T, G, and C contents of the complete mtDNA of Cy. catinatum, Cs. minutus, P. imparidentatum ranged from 29.79–30.59%, 44.86–45.53%, 16.66–17.99%, and 7.22–7.37%, respectively. The detailed annotations of the three mt genomes, including the position and length of each gene, A+T content, initiation codon, and termination codon of 12 protein-coding genes, are listed in Table 2.
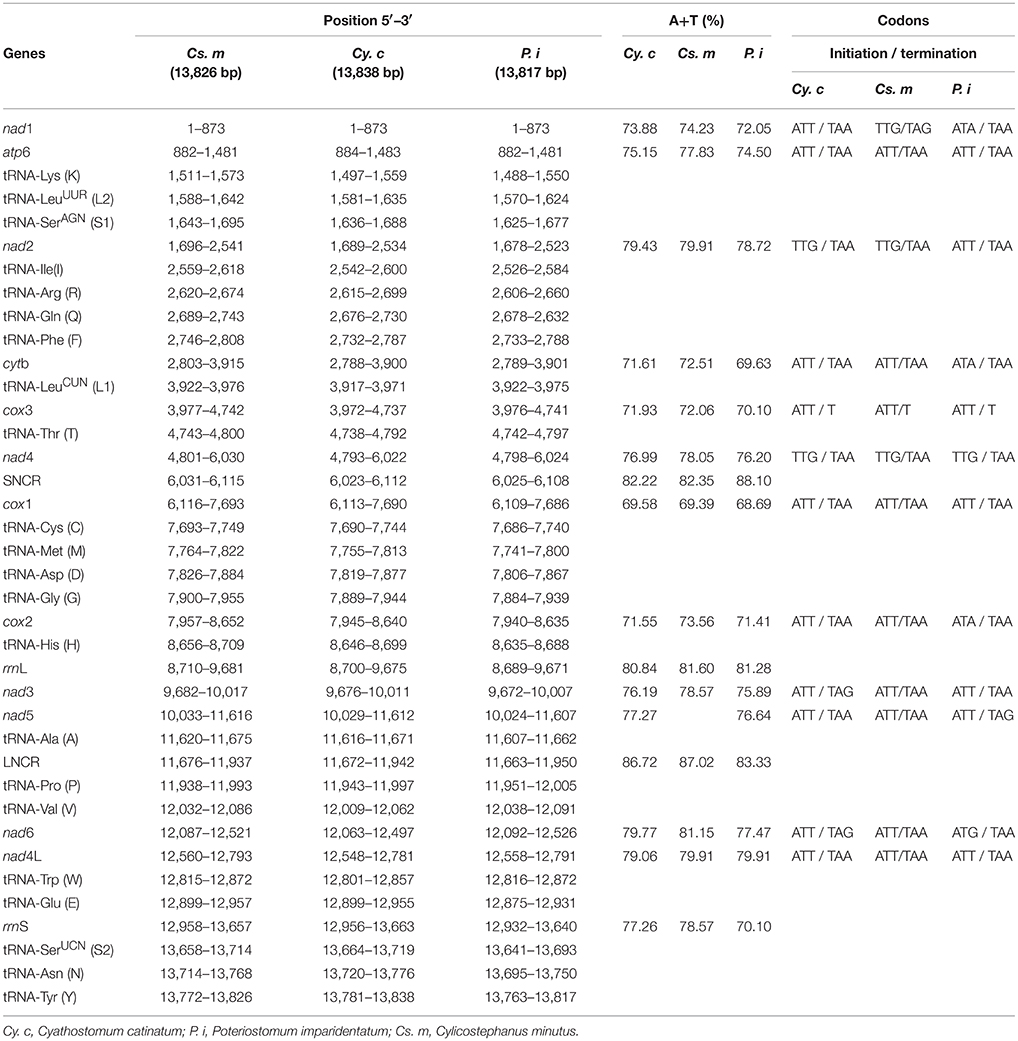
Table 2. Mitochondrial genome organization of three nematodes.
Comparative Analysis
Homology analysis showed that the highest identity (91.6%) occurred between the mt genome of Cy. catinatum and that of Cs. goldi (Table 3). Compared with the three nematodes of Strongylus (S. equinus and S. vulgaris) and Macropicola (M. ocydromi) in the same subfamily (Strongylinae), the worms in Cyathostominae showed relatively higher similarities to Triodontophorus species. Furthermore, the results from these analyses indicate that the mt genome arrangements of the Strongyloidea nematodes sequenced to date are identical. The detailed comparison is listed in Tables 2–4.
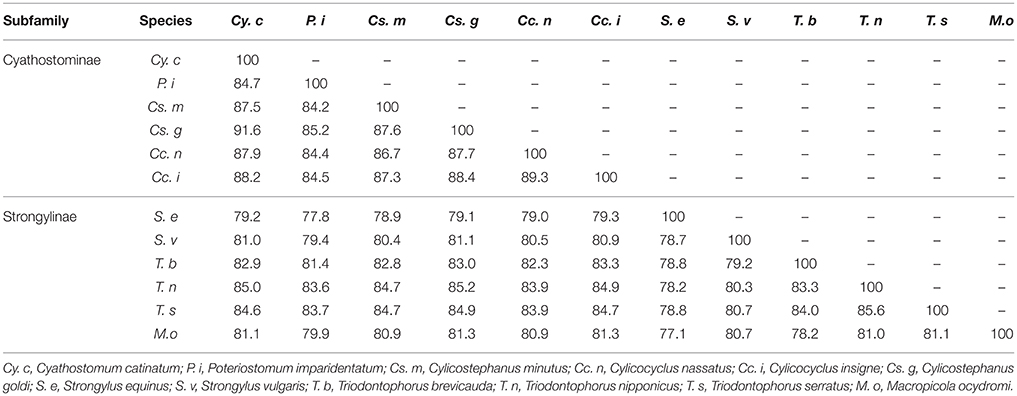
Table 3. The comparative analysis of mt DNA among the family Strongylidae.
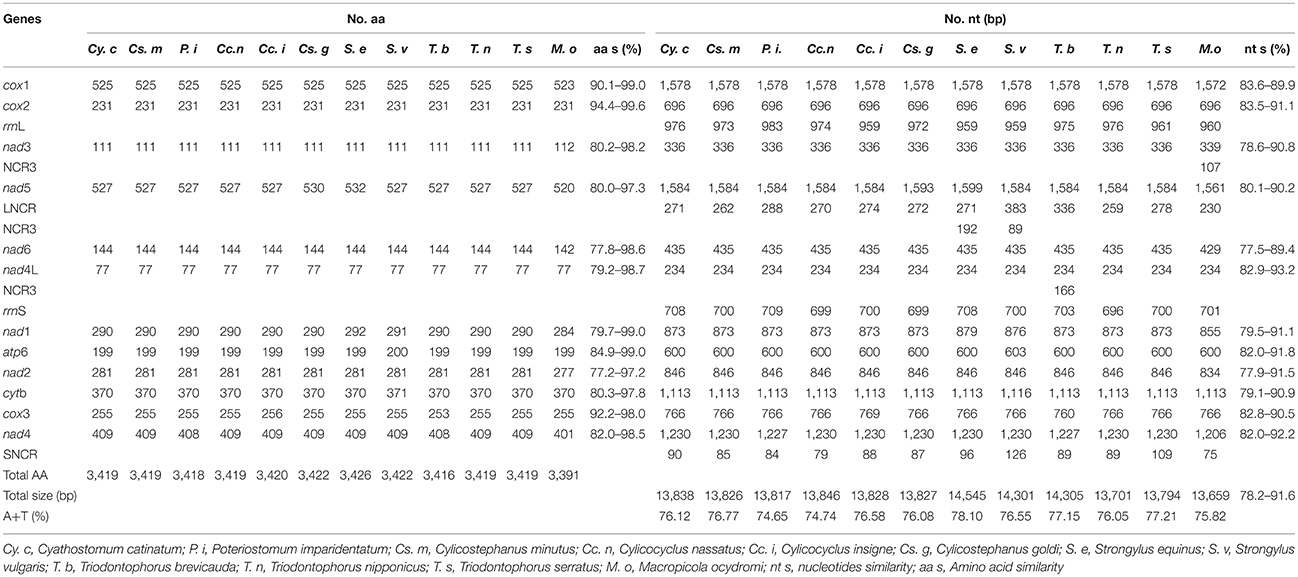
Table 4. The complete nucleotide identify analyses of 12 nematodes in the family Strongylidae.
Comparison of the 12 protein-coding genes of all Strongylidae nematodes revealed that the nad4L gene is the most conserved gene in terms of nucleotide sequences; however, the cox2 gene is the most conserved at the amino acid level (Table 4). For the codon usage of the 12 protein-coding genes of the three worms analyzed in this study, ATT initiation codons were present in high frequency, TAA was the most shared termination codon, and incomplete codon T appeared at the end of the cox3 gene of the three nematodes.
Phylogenetic Analysis
In the present study, mtDNA data were employed to assess the phylogenetic relationships of Strongyloidea nematodes, and the results are presented in Figure 1. All phylogenetic trees generated similar topologies, and the horse-parasitizing nematodes always clustered together in each tree. However, phylogenetic analysis using different methods revealed different relative positions for some species, such as P. imparidentatum. MP and ML trees showed the same topology, except for the branch that contained O. columbianum (Figures 1B,C). Congeneric species always formed clades (T. brevicauda, T. nipponicus, and T. serratus; Cc. insigne and Cc. nassatus; and Ch. erschowi and Ch. ovina), and the genera Oesophagostomum and Cylicostephanus are polyphyletic. Cs. goldi and Cy. catinatum formed sister taxa in all three trees. P. imparidentatum was sister to a clade composed of Cc. insigne, Cs. goldi, Cc. nassatus, Cy. catinatum, T. brevicauda, T. nipponicus, and T. serratus in the BI tree. However, P. imparidentatum clustered with all Cyathostominae nematodes in the MP and ML trees. Triodontophorus species clustered together with all the Cyathostominae nematodes with strong support in the three phylogenetic trees. In particular, in the BI tree, the Triodontophorus species clustered with Cylicostephanus, Cyathostomum, Cylicocyclus, and Poteriostomum within the clade of Cyathostominae. The two Strongylus species formed sister groups in the ML and MP trees, but not in the BI tree. The kangaroo-parasitizing nematodes (H. macropi and M. ocydromi) formed a distinct branch with the worms of cattle, sheep, goats, and pigs (C. erschowi, C. ovina, O. asperum, O. columbianum, O. dentatum, O. quadrispinulatum), even though M. ocydromi is classified in Strongylinae. The Oesophagostomum species did not form a monophyletic group, because of H. macropi, M. ocydromi, and the Chabertia species. However, O. quadrispinulatum and O. dentatum formed a distinct branch, and O. asperum was sister to the Chabertia species in all three trees. Syngamus trachea was divergent from all other species in the three analyses (see Figure 1).
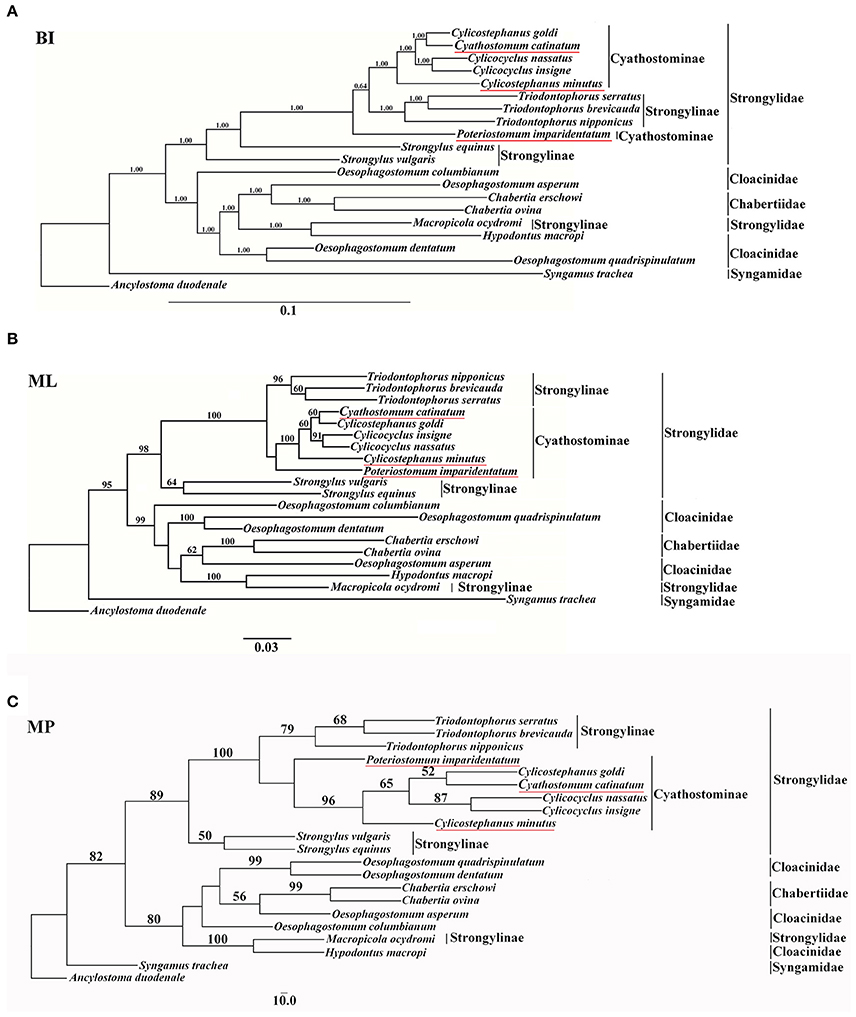
Figure 1. Phylogenetic relationships of 20 Strongyloidea nematodes based on concatenated amino acid sequences of 12 protein-coding genes were analyzed by Bayesian inference (BI) (A), maximum parsimony (MP) (C), and maximum likelihood (ML) (B) using Ancylostoma duodenale as an outgroup. The values below 50% are not shown.
Discussion
The mitochondrial genome is important in molecular biology, and is extensively applied when studying the taxonomy, population genetics, and systematics of parasites (Gissi et al., 2008; Zhang et al., 2015; Guo et al., 2016; Liu et al., 2016). In the present study, we obtained the mtDNA of three Cyathostominae nematodes for the first time and compared these mtDNA sequences those of Strongylidae species. The complete mtDNA sequences of the three nematodes were slightly different in length compared with the other Strongylidae nematodes (except for S. equinus and T. brevicauda), which is consistent with the length of typical metazoan mt genomes (Gissi et al., 2008; Jia et al., 2010; Tian et al., 2015; Zhang et al., 2015; Guo et al., 2016; Liu et al., 2016). Because of the longer length of the non-coding regions, the S. equinus and T. brevicauda mt genomes were slightly longer than other Strongylidae nematodes mt genomes. This phenomenon is also found in the mt genomes of entomopathogenic nematodes and plant-parasitic nematodes. For example, the lengths of the complete mt genomes of the entomopathogenic nematode Steinernema litorale and the plant-parasitic nematode Pratylenchus vulnus are 21,403 bp and 21,656 bp in size, respectively; however, the size of the non-coding regions was extremely long (S. litorale, 8,137 bp; P. vulnus, 7,748 bp) (Sultana et al., 2013; Taisei et al., 2016). The total A+T contents of three worms mtDNA of this study were consistent with those of most nematode mt genomes characterized to date, such as T. brevicauda (77.0%) and Wuchereria bancrofti (74.6%) (Ramesh et al., 2012; Duan et al., 2015), whereas the percentages of A+T content are remarkably higher than those of trematodes and protozoa, such as Echinostoma hortense (63.03%) and Eimeria magna (65.16%) (Tian et al., 2015; Liu et al., 2016). The A+T content in non-coding regions was higher than those in other regions of the mt genomes, which is consistent with the mt genomes of other nematodes in previous studies (Hu et al., 2002; Lin et al., 2012; Ramesh et al., 2012). However, non-coding regions quantity differed from those of other nematodes in the same family, and the third non-coding region was found in the mt genomes of T. brevicauda, Strongylus equinus, and M. ocydromi. There are also several pairs of repeats in the long non-coding regions of the three nematodes in this study with 12 bp for direct repeats and 12–14 bp for inverted repeats. This phenomenon was also found in the long non-coding regions of other helminthes, such as Strongyloides stercoralis, W. bancrofti, Fascioloides magna, Taenia multiceps, T. hydatigena, and T. pisiformis, but the lengths of those repeat sequences were longer than those in the three worms in the present study (Hu et al., 2003; Jia et al., 2010; Ramesh et al., 2012; Ma et al., 2016). Nevertheless, the functions of these regions remain unclear.
The mt genome of the three nematodes was encoded on the same strand and transcribed in the same direction, which is consistent with those of other Chromadorea nematodes available in GenBank, but different from those of Enoplea nematodes, such as Trichinella spiralis, Trichuris trichiura, and T. ovis (Lavrov and Brown, 2001; Liu et al., 2012a,b). Interestingly, the gene order in this study is also identical to that of 17 species that belong to superfamily Strongyloidea, for which mt genomes are available in GenBank, including Strongylidae (Cs. goldi, Cc. insigne, Cc. nassatus, S. equinus, S. vulgaris, T. brevicauda, T. nipponicus, T. serratus, and M. ocydromi), Chabertiidae (C. erschowi and C. ovina), Cloacinidae (O. asperum, O. columbianum, O. dentatum, O. quadrispinulatum, and H. macropi), and Syngamidae (Syngamus trachea). This phenomenon was also found in another group of the family Trichuridae, including Trichuris trichiura, T. suis, T. ovis, and T. discolor, whose mt genomes have the same gene arrangement. However, in other nematodes, such as the family Oxyuridae, Syphacia obvelata mt genome gene arrangement is consistent with that of Wellcomia siamensis, but different from those of Enterobius vermicularis and Aspiculuris tetraptera (Wang et al., 2016).
The results of the present study showed that topologies of the three trees with different phylogenetic reconstruction strategies were identical or similar. The paraphyly of Oesophagostomum species and Cylicostephanus species caused by Chabertia species and Cy. catinatum was evident in the mtDNA analyses. These paraphyletic groups were also proposed by some previous studies based on rDNA sequence (Hung et al., 2000; Gouý de Bellocq et al., 2001). P. imparidentatum was evolutionarily distant from other Cyathostominae and Triodontophorus species. Additionally, P. imparidentatum displayed the lowest nucleotide identity when compared with the other nematodes (Cy. catinatum, Cc. nassatus, Cc. insigne, Cs. minutus, Cs. goldi, T. brevicauda, T. nipponicus, and T. serratus). The phylogenetic analysis indicated that subfamily Cyathostominae was closer to the Triodontophorus species that belong to Strongylinae than to the Strongylus species that belong to Strongylinae.
This phenomenon was also reflected in the complete nucleotide identity comparisons; the Strongylus species showed relatively low nucleotide identity to the sequences of other equine strongyles. Nevertheless, the Triodontophorus worms mtDNA sequences have higher identity with the Cyathostominae than that of Strongylus species, which is consistent with the phylogenetic results. The similarity results were also further supported by other studies. The mtDNA-based phylogenetic trees revealed that all the worms in Cyathostominae, T. serratus, and Craterostomum acuticaudatum clustered with each other rather than the three other nematodes of Strongylinae (S. equinus, S. vulgaris, S. edentatus) (McDonnell et al., 2000). Another phylogenetic analysis of the large subunit rDNA D3 domain for 21 species of equine strongyles showed that T. serratus and T. brevicauda clustered with Cyathostominae rather than Strongylinae (Zhang et al., 2007). Furthermore, there is also biological evidence that supports this relationship; for example, a comparative study of the morphology of the L4s of 20 equine strongyles revealed that the larvae of Triodontophorus were more similar to those of Cyathostominae than to those of Strongylus species (Dvojnos and Kharchenko, 1990).
The phylogenetic relationships based on the complete mt genomes in this study indicated that the relationships among equine strongyles are inconsistent with the traditional classification. Triodontophorus species are genetically different from Strongylus species, and are more closely related to Cyathostominae. These analyses supported our hypothesis that Triodontophorus species belong to Cyathostominae.
Conclusions
The findings of the present study, which used comparative and phylogenetic analyses of mtDNA sequences, supported the hypothesis that Triodontophorus species belong to Cyathostominae. The availability of the complete mt genome sequences of Cy. catinatum, Cs. minutus, and P. imparidentatum can provide novel genetic markers for further studies on the taxonomy, population genetics, and systematics of Strongylidae nematodes.
Data Availability Statement
Representative nucleotide sequences were deposited in GenBank with the following accession numbers: KY495600-KY495602 for the mtDNA, and KY495603-KY495605 for the ITS.
Author Contributions
CRW conceived and designed the study, and critically revised the manuscript. YG, HD, and WWX performed the experiments. YG and QCC analyzed the data. YG drafted the manuscript. YZ, XY, and JHQ helped in study design, study implementation and manuscript preparation. All authors read and approved the final manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank the staff and workers in the abattoir who helped in the collection of nematodes. This work was supported by grant from the National Key Research and Development Program of China (2017YFD0501300), the grant of Heilongjiang Bayi Agricultural University Graduate Innovative Research Project (YJSCX2017-Y37), and the International Science & Technology Cooperation Program of China (Grant No. 2013DFA31840).
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmicb.2017.01444/full#supplementary-material
Table S1. The primers used for obtain mitochondrial genomes of the three nematodes.
Figure S1. The detail information of morphological characteristics of Cyathostomum catinatum. (A) The head of Cy. catinatum, (B) The tail of female, (C) The tail of male, Bar = 0.1 mm.
Figure S2. The detail information of morphological characteristics of Cylicostephanus minutus. (A) The head of Cs. minutus, (B) The tail of female, (C) The tail of male, Bar = 0.1 mm.
Figure S3. The detail information of morphological characteristics of Poteriostomum imparidentatum. (A) The head of P. imparidentatum, (B) The tail of female, (C) The tail of male, Bar = 0.1 mm.
References
Bu, Y., Niu, H., Gasser, R. B., Beveridge, I., and Zhang, L. (2009). Strongyloid nematodes in the caeca of donkeys in Henan Province, China. Acta Parasitol. 54, 263–268. doi: 10.2478/s11686-009-0037-z
Burland, T. G. (2000). DNASTAR's Lasergene sequence analysis software. Methods Mol. Biol. 132, 71–91. doi: 10.1385/1-59259-192-2:71
Corning, S. (2009). Equine cyathostomins: a review of biology, clinical significance and therapy. Parasit. Vectors 2:S1. doi: 10.1186/1756-3305-2-S2-S1
Duan, H., Gao, J. F., Hou, M. R., Zhang, Y., Liu, Z. X., Gao, D. Z., et al. (2015). Complete Mitochondrial genome of an equine intestinal parasite, Triodontophorus brevicauda (Chromadorea: Strongylidae): the first characterization within the genus. Parasitol. Int. 64, 429–434. doi: 10.1016/j.parint.2015.06.006
Durette-Desset, M. C., Beveridge, I., and Spratt, D. M. (1994). The origins and evolutionary expansion of the Strongylida (Nematoda). Int. J. Parasitol. 24, 1139–1165.
Dvojnos, G. M., and Kharchenko, V. A. (1990). Morphology and differential diagnostics of parasitic larvae of some Strongylidae (Nematoda) of horses. Angew. Parasitol. 31, 15–28.
Gao, Y., Qiu, J. H., Zhang, B. B., Su, X., Fu, X., Yue, D. M., et al. (2017). Complete mitochondrial genome of parasitic nematode Cylicocyclus nassatus, and comparative analyses with Cylicocyclus insigne. Exp. Parasitol. 172, 18–22. doi: 10.1016/j.exppara.2016.11.005
Gasser, R. B., Bott, N. J., Chilton, N. B., Hunt, P., and Beveridge, I. (2008). Toward practical, DNA-based diagnostic methods for parasitic nematodes of livestock-bionomic and biotechnological implications. Biotechnol. Adv. 26, 325–334. doi: 10.1016/j.biotechadv.2008.03.003
Gissi, C., Iannelli, F., and Pesole, G. (2008). Evolution of the mitochondrial genome of Metazoa as exemplified by comparison of congeneric species. Heredity 101, 301–320. doi: 10.1038/hdy.2008.62
Gouý de Bellocq, G., Ferte, H., Depaquit, J., Justine, J. L., Tillier, A., and Durette-Desset, M. -C. (2001). Phylogeny of the Trichostrongylina (Nematoda) inferred from 28S rDNA sequences. Mol. Phylogenet. Evol. 19, 430–442. doi: 10.1006/mpev.2001.0925
Guindon, S., and Gascuel, O. (2003). A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst. Biol. 52, 696–704. doi: 10.1080/10635150390235520
Guo, D. H., Zhang, Y., Fu, X., Gao, Y., Liu, Y. T., Qiu, J. H., et al. (2016). Complete mitochondrial genomes of Dermacentor silvarum and comparative analyses with another hard tick Dermacentor nitens. Exp. Parasitol. 169, 22–27. doi: 10.1016/j.exppara.2016.07.004
Hu, M., Chilton, N. B., and Gasser, R. B. (2002). The mitochondrial genomes of the human hookworms, Ancylostoma duodenale and Necator americanus (Nematoda: Secernentea). Int. J. Parasitol. 32, 145–158. doi: 10.1016/S0020-7519(01)00316-2
Hu, M., Chilton, N. B., and Gasser, R. B. (2003). The mitochondrial genome of Strongyloides stercoralis (Nematoda) - idiosyncratic gene order and evolutionary implications. Int. J. Parasitol. 33, 1393–1408. doi: 10.1016/S0020-7519(03)00130-9
Hung, G., Chilton, N. I., and Gasser, R. (2000). A molecular systematic framework for equine strongyles based on ribosomal DNA sequence data. Int. J. Parasitol. 30, 95–103. doi: 10.1016/S0020-7519(99)00166-6
Jia, W. Z., Yan, H. B., Guo, A. J., Zhu, X. Q., Wang, Y. C., Shi, W. G., et al. (2010). Complete mitochondrial genomes of Taenia multiceps, T. hydatigena and T. pisiformis: additional molecular markers for a tapeworm genus of human and animal health significance. BMC Genomics 11:447. doi: 10.1186/1471-2164-11-447
Lavrov, D. V., and Brown, W. M. (2001). Trichinella spiralis mtDNA: A nematode mitochondrial genome that encodes a putative ATP8 and normally structured tRNAs and has a gene arrangement relatable to those of coelomate metazoans. Genetics 157, 621–637.
Lichtenfels, J. R., Kharchenko, V. A., and Dvojnos, G. M. (2008). Illustrated identification keys to strongylid parasites (Strongylidae: Nematoda) of horses, zebras and asses (Equidae). Vet. Parasitol. 156, 4–161. doi: 10.1016/j.vetpar.2008.04.026
Lichtenfels, J. R., Kharchenko, V. A., Krecek, R. C., and Gibbons, L. M. (1998). An annotated checklist by genus and species of 93 species level names for 51 recognized species of small strongyles (Nematoda, Strongyloidea, Cyathostominea) of horses, asses and zebras of the world. Vet. Parasitol. 79, 65–79.
Lin, R. Q., Liu, G. H., Zhang, Y., D'Amelio, S., Zhou, D. H., Yuan, Z. G., et al. (2012). Contracaecum rudolphii B: gene content, arrangement and composition of its complete mitochondrial genome compared with Anisakis simplex s.l. Exp. Parasitol. 130, 135–140. doi: 10.1016/j.exppara.2011.11.003
Liu, G. H., Gasser, R. B., Su, A., Nejsum, P., Peng, L., Lin, R. Q., et al. (2012a). Clear genetic distinctiveness between human- and pig-derived trichuris based on analyses of mitochondrial datasets. PLoS Negl. Trop. Dis. 6:e1539. doi: 10.1371/journal.pntd.0001539
Liu, G. H., Wang, Y., Xu, M. J., Zhou, D. H., Ye, Y. G., Li, J. Y., et al. (2012b). Characterization of the complete mitochondrial genomes of two whipworms Trichuris ovis and Trichuris discolor (Nematoda: Trichuridae). Infect. Genet. Evol. 12, 1635–1641. doi: 10.1016/j.meegid.2012.08.005
Liu, G. H., Yan, H. B., Otranto, D., Wang, X. Y., Zhao, G. H., Jia, W. Z., et al. (2014). Dicrocoelium chinensis and Dicrocoelium dendriticum (Trematoda: Digenea) are distinct lancet fluke species based on mitochondrial and nuclear ribosomal DNA sequences. Mol. Phylogenet. Evol. 79, 325–331. doi: 10.1016/j.ympev.2014.07.002
Liu, Z. X., Zhang, Y., Liu, Y. T., Chang, Q. C., Su, X., Fu, X., et al. (2016). Complete Mitochondrial Genome of Echinostoma hortense (Digenea: Echinostomatidae). Korean J. Parasitol. 54, 173–179. doi: 10.3347/kjp.2016.54.2.173
Love, S., Murphy, D., and Mellor, D. (1999). Pathogenicity of cyathostome infections. Vet. Parasitol. 85, 113–122.
Lyons, E. T., Kuzmina, T. A., and Tolliver, S. C. (2011). Observations on development of natural infection and species composition of small strongyles in young equids in Kentucky. Parasitol. Res. 109, 1529–1535. doi: 10.1007/s00436-011-2460-y
Ma, J., He, J. J., Liu, G. H., Leontovyč, R., Kašný, M., and Zhu, X. Q. (2016). Complete mitochondrial genome of the giant liver fluke Fascioloides magna (Digenea: Fasciolidae) and its comparison with selected trematodes. Parasit. Vectors 9:429. doi: 10.1186/s13071-016-1699-7
McDonnell, A., Love, S., Tait, A., Lichtenfels, J. R., and Matthews, J. B. (2000). Phylogenetic analysis of partial mitochondrial cytochrome oxidase c subunit I and large ribosomal RNA sequences and nuclear internal transcribed spacer I sequences from species of Cyathostominae and Strongylinae (Nematoda, Order Strongylida), parasites of the horse. Parasitology 121, 649–659. doi: 10.1017/S003118200000696X
Morariu, S., Mederle, N., Badea, C., Dărăbuş, G., Ferrari, N., and Genchi, C. (2016). The prevalence, abundance and distribution of cyathostomins (small stongyles) in horses from Western Romania. Vet. Parasitol. 223, 205–209. doi: 10.1016/j.vetpar.2016.04.021
Mughini, G. L., Usai, F., and Stancampiano, L. (2011). Strongylosis in horses slaughtered in Italy for meat production: Epidemiology, influence of the horse origin and evidence of parasite self-regulation. Vet. Parasitol. 179, 167–174. doi: 10.1016/j.vetpar.2011.02.024
Page, R. D. (1996). TreeView: an application to display phylogenetic trees on personal computers. Comput. Appl. Biosci. 12, 357–358.
Ramesh, A., Small, S. T., Kloos, Z. A., Kazura, J. W., Nutman, T. B., Serre, D., et al. (2012). The complete mitochondrial genome sequence of the filarial nematode Wuchereria bancrofti from three geographic isolates provides evidence of complex demographic history. Mol. Biochem. Parasitol. 183, 32–41. doi: 10.1016/j.molbiopara.2012.01.004
Reinemeyer, C. R. (1986). Small strongyles. Recent advances. Vet. Clin. N. Am. Eq. Pract. 2, 281–312.
Ronquist, F., and Huelsenbeck, J. P. (2003). MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19, 1572–1574. doi: 10.1093/bioinformatics/btg180
Singh, G., Singh, N. K., Singh, H., and Rath, S. S. (2016). Assessment of risk factors associated with prevalence of strongyle infection in equines from Central Plain Zone, Punjab. J. Parasit. Dis. 40, 1381–1385. doi: 10.1007/s12639-015-0695-8
Sultana, T., Kim, J., Lee, S. H., Han, H., Kim, S., Min, G. S., et al. (2013). Comparative analysis of complete mitochondrial genome sequences confirms independent origins of plant-parasitic nematodes. BMC Evol. Biol. 13:12. doi: 10.1186/1471-2148-13-12
Swofford, D. L. (2002). PAUP*. Phylogenetic Analysis Using Parsimony (* and Other Methods). Sunderland, MA: Sinauer Associates.
Taisei, K., Tanzila, A., and Mutsuhiro, Y. (2016). Complete mitochondrial genomes of four entomopathogenic nematode species of the genus Steinernema. Parasit. Vectors 9, 430. doi: 10.1186/s13071-016-1730-z
Tamura, K., Peterson, D., Peterson, N., Stecher, G., Nei, M., and Kumar, S. (2011). MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28, 2731–2739. doi: 10.1093/molbev/msr121
Thompson, J. D., Gibson, T. J., Plewniak, F., Jeanmougin, F., and Higgins, D. G. (1997). The Clustal X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic. Acids Res. 25, 4876–4882.
Tian, S. Q., Cui, P., Fang, S. F., Liu, G. H., Wang, C. R., and Zhu, X. Q. (2015). The complete mitochondrial genome sequence of Eimeria magna (Apicomplexa: Coccidia). Mitochondrial DNA 26, 714–715. doi: 10.3109/19401736.2013.843088
Traversa, D., Milillo, P., Barnes, H., von Samson-Himmelstjerna, G., Schurmann, S., Demeler, J., et al. (2010). Distribution and species-specific occurrence of cyathostomins (Nematoda, Strongylida) in naturally infected horses from Italy, United Kingdom and Germany. Vet. Parasitol. 168, 84–92. doi: 10.1016/j.vetpar.2009.10.006
Wang, C. R., Lou, Y., Gao, J. F., Qiu, J. H., Zhang, Y., Gao, Y., et al. (2016). Comparative analyses of the complete mitochondrial genomes of the two murine pinworms Aspiculuris tetraptera and Syphacia obvelata. Gene 585, 71–75. doi: 10.1016/j.gene.2016.03.037
Wang, Y., Wang, C. R., Zhao, G. H., Gao, J. F., Li, M. W., and Zhu, X. Q. (2011). The complete mitochondrial genome of Orientobilharzia turkestanicum supports its affinity with African Schistosoma spp. Infect. Genet. Evol. 11, 1964–1970. doi: 10.1016/j.meegid.2011.08.030
Zhang, L., Hu, M., Chilton, N. B., Huby-Chilton, F., Beveridge, I., and Gasser, R. B. (2007). Nucleotide alterations in the D3 domain of the large subunit of ribosomal DNA among 21 species of equine strongyle. Mol. Cell. Probes 21, 111–115. doi: 10.1016/j.mcp.2006.08.008
Keywords: complete mitochondrial genome, Strongyloidea, Strongylidae, Cyathostominae, Strongylinae, phylogenetic analysis
Citation: Gao Y, Zhang Y, Yang X, Qiu J-H, Duan H, Xu W-W, Chang Q-C and Wang C-R (2017) Mitochondrial DNA Evidence Supports the Hypothesis that Triodontophorus Species Belong to Cyathostominae. Front. Microbiol. 8:1444. doi: 10.3389/fmicb.2017.01444
Received: 29 April 2017; Accepted: 17 July 2017;
Published: 03 August 2017.
Edited by:
Guan Zhu, Texas A&M University, United StatesReviewed by:
Mario Santoro, Istituto Zooprofilattico Sperimentale del Mezzogiorno, ItalyGuanghui Zhao, Northwest A&F University, China
Copyright © 2017 Gao, Zhang, Yang, Qiu, Duan, Xu, Chang and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qiao-Cheng Chang, changqiaocheng2001@163.com
Chun-Ren Wang, chunrenwang@sohu.com